Căutare
Căutare
Toate procedurile
Analiza detaliată
Analiza detaliată
Aniridia congenitală este caracterizată prin absența totală sau parțială a irisului și a fost descrisă pentru prima oară în 1819 de către Baratta. În greacă, ”aniridia” înseamnă ”fără iris”. Studiile epidemiologice arată o frecvență de 118 cazuri la 7,6 milioane de locuitori, ceea ce indică o incidență de un caz la 56.000 de nașteri cu nou – născuți vii. Există o ușoară preponderență la sexul masculin, comparativ cu sexul feminin.
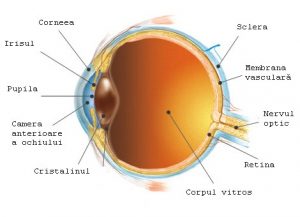 Globul ocular este alcătuit din punct de vedere anatomic din trei straturi și conținut, după cum urmează:
Globul ocular este alcătuit din punct de vedere anatomic din trei straturi și conținut, după cum urmează:
Irisul este situat în faţa cristalinului și prezintă o față anterioară formată dintr-o zonă ciliară cu aspect striat radiar dat de traiectul vaselor iriene stromale și o zonă pupilară cu aspect neted. Această față a irisului delimitează camera anterioară a ochiului, cuprinsă între faţa posterioară a corneei, faţa anterioară diafragmului irido-cristalinian şi extern unghiul camerular. Fața posterioară a irisului delimitează camera posterioară a ochiului, cuprinsă anterior de iris, posterior de cristalin şi zonula lui Zinn, iar extern de procesele ciliare. Cele două zone ale irisului sunt separate la nivel colaretului irian care este o proeminență festonată, vizibilă sau nu in funcție de cantitatea de pigment. Fața anterioară are o colorație variabilă de la un individ la altul in funcție de numarul de melanocite din stroma iriană.
Anexele globului ocular sunt reprezentate de urmatoarele structuri: pleoape, conjunctiva, aparatul lacrimal, sistemul muscular extrinsec, orbita.
După modul de transmitere genetică, aniridia se clasifică în trei tipuri genetice și anume:
Genele implicate în producerea aniridiei fac parte din familia unor gene numite PAX. Gena PAX 6 (paired box 6), exprimată la nivelul diencefalului, este considerată o genă „master”, implicată în dezvoltarea globilor oculari localizată pe cromozomul 11p13. De asemenea, s- a demonstrat faptul că această genă are rol și în dezvoltarea neurologică normală, iar exprimarea ei este importantă în maturarea irisului și a corpului ciliar. Până în prezent, au fost identificate mai mult de 400 de variante heterozigote ale genei PAX 6. Cele mai multe dintre mutațiile heterozigote ale genei PAX 6, includ inserții, deleții, mutații non-sens și de matisare a exonilor introducerea de codoni stop prematuri în zona de citire a genei PAX 6, avâd ca rezultat haploinsuficiența genei. Nivelul expresiei proteinei produsă de o singură alelă funcțională nu este suficient pentru a produce o proteină PAX 6 biologic activă.
Aniridia ca și afectare oculară nu se limitează doar la nivelul irisului, ci poate avea efecte și asupra corneei, foveei, nervului optic sau cristalinului. Fenotipul variază de la o familie la alta și de la un membru al familiei la celălalt. Adesea, bebelușii cu aniridie sunt îndrumați către medicul oftalmolog datorită pupilelor fixate in dilatație sau datorită nistagmusului secundar. Persoanele cu aniridie prezintă în mod caracteristic, nistagmus, acuitate vizuală scăzută (de obicei, 20 de cazuri 100 – 200 copii) și hipoplazie foveală. Acuitatea vizuală redusă este datorată ambliopiei, edemului cornean după operația de cataractă sau degenerării maculare legate de vârstă. Forme mai ușoare de aniridia cu schimbări de arhitectură subtilă a irisului pot avea o acuitate viziuală bună și o structură foveală normală.
Reducerea acuității vizuale este cauzată în primul rând de hipoplazia foveală, dar cataracta, glaucomul și opacifierea corneei sunt responsabile de eșecul vizual progresiv. Majoritatea copiilor cu aniridie prezintă anomalii evidente ale irisului si ale pupilei, iar in jurul vârstei de șase săptămâni pot dezvolta nistagmus.
Efectele aniridiei asupra irisului – cea mai comună manifestare a aniridiei este lipsa totală sau parțială a irisului. În alte cazuri, irisul se găsește sub forma unui mic țesut rezidual vizibil la gonioscopie sau biomicroscopia cu ultrasunete. Uneori, dimensiunea irisului poate fi normală, dar poate exista o pierdere a arhitecturii suprafeței irisului.
Efecte asupra presiunii intraoculare: două treimi dintre pacienți pot dezvolta glaucom cu presiune intraoculară ridicată. Presiunea intraoculară crescută este asociată cu pierderea celulelor ganglionare retiniene care determină reducerea câmpului vizual. Incidența glaucomului în cadrul aniridiei este de 10% până la 75% și poate să apară in timpul preadolescenței sau la adultul tânăr. Glaucomul este principala cauză de pierdere a vederii în aniridie, de aceea pacienții trebuie monitorizați în această privință la intervale regulate.
În aniridie, se bănuiește că glaucomul se formează din cauza anomaliilor de dezvoltare în unghiul de drenaj al ochiului, care împiedică scurgerea umoarei apoase prin canalul lui Schlemm. Diagnosticul implică examinarea unghiului pentru evidențierea închiderii, măsurarea presiunii intraoculare, examinarea nervului optic și evaluarea câmpului vizual. Pacienții cu aniridie au cornee de până la 100 μm mai groasă decât media, ceea ce face ca tonometria să nu fie fiabilă la acești pacienți atât în diagnosticul cât și în managementul glaucomului aniridic.
Efecte asupra nervului optic: hipoplazia nervului optic poate să apară până la 10% din cazuri și poate asocia coloboame de nerv optic.
Efecte asupra fovee: hipoplazia foveală se caracterizează prin hipopigmentare maculară și reflex foveal redus, adesea abolit.
Printre tulburările de refracție ce se găsesc in cadrul aniridiei putem întâlni miopie, hipermetropie, astigmatism, iar 10% dintre pacienți pot prezenta ptoză palpebrală. Rezultatele unui studiu realizat pe 35 de subiecți referitor la frecvența complicațiilor apărute la pacienții cu aniridie sunt prezentate în tabelul de mai jos.
Frecvența complicațiilor aniridiei |
|
| Manifestarea clinică sau complicația | Frecvența cu care apar |
| Nistagmus | 76% |
| Cataracta | 56% |
| Glaucom | 64% |
| Keratopatie | 48% |
| Hipoplazie foveală | 20% |
| Hipoplazia nervului optic | 20% |
| Presiunea intraoculară crescută | 36% |
Aniridia este, de obicei, diagnosticată pentru prima dată din punct de vedere clinic de către medicul pediatru. Elementele clinice care atrag atenția încă de la început asupra tabloului clinic de aniridie pot fi amintite astfel:
Cele mai potrivite metode de explorare clinică a aniridiei și complicațiilor sale sunt urmatoarele:
În urma diagnosticului clinic de aniridie este important să se evalueze antecedentele familiale. Chiar și fără antecedente familiale evidente, examinarea oftalmologică a părinților ar trebui efectuată pentru spectrul de anomalii al genei PAX 6.
În analiza moleculară a cazurilor de aniridie, primul pas este de a determina dacă există o deleție subiacentă care implică ambele gene PAX6 și WT1 (care determină sindromul WAGR și care predispune la tumora Wilms). În cazurile în care este decelată existența mutației genei WT1 consilierea genetică este deosebit de importantă, detectarea unei mutații PAX6 fiind mai puțin urgentă.
Descoperirea din primele stadii ale tumorii Wilms este foarte impotantă datorită răspunsului favorabil al acesteia la tratament. Dacă nu există istoric familial, nefiind identificată o mutație la părinți, atunci concluzia este că s-a produs o mutație de novo, însă nu poate fi exclusă existența unui mozaic germinal la părinți, în acest caz, riscul de recurență fiind de 1 – 2%.
Majoritatea pacienților diagnosticați cu aniridie izolată au un părinte afectat. Cei care nu au istoric familial pot avea o mutație sau o deleție de novo a genei PAX 6. Fenotipul poate varia foarte mult între membrii familiei, astfel încât părinții oricărei persoane cu mutație aparentă de novo ar trebui să fie examinați pentru modificări ale genei PAX 6. 70% dintre persoanele cu aniridie izolată au un părinte afectat iar 30% au o variantă patogenă de novo a genei PAX 6 sau o deleție a unei regiuni de reglare care controlează expresia genei PAX 6. Fiecare copil al unui individ cu aniridia izolată are un risc de 50% de a moșteni modificarea genetică cauzală și de a dezvolta aniridia. Diagnosticul prenatal prin amniocenteză (15 – 18 săptămâni de gestație) sau prelevarea de biopsie de vilozități corionice (10 – 12 săptămâni de gestație) este posibilă pentru:
Este esențial să se facă o diagnosticare precoce și corectă a aniridiei pentru a determina dacă pacientul are o mutație a genei PAX 6 sau dacă el sau ea poartă o deleție care implică gena WT1, care determină apariția tumorii Wilms. Toți pacienții cu aniridia non – familială trebuie testați pentru prezența unei mutații ale genei PAX 6 sau a unei deleții de cromozomului 11p13, deoarece unii pacienți prezintă riscul de a dezvolta tumora Wilms.
Un pacient cu absența aproape totală a irisului, prezența nistagmusului și a hipoplaziei foveale, nu pune mari probleme de diagnostic. Totuși, aniridia se diferențiază de anumite patologii: anomaliile de dezvoltare anterioară ale segmentului ( sindromul Axenfeld-Rieger și anomalia Peters), albinismul (oculocutanat și ocular) și alte cauze ale nistagmusului infantil. Se impune de asemenea diagnostic diferențial și cauzele de diminuare a acuității vizuale fără modificări ale irisului, cum ar fi displazia retiniană, distrofie, cataractă congenitală, hipoplazie nervoasă optică și infecții congenitale.
Cauzele absenței parțiale sau complete a irisului la adulți includ traumatisme, intervenții chirurgicale oculare anterioare și sindroame iridocorneale. Vârsta la debut, antecedentele personale medicale și absența altor caracteristici oculare împiedică confuzia diagnosticului cu aniridia congenitală.
Pentru a stabili amploarea bolii și a nevoilor unei persoane diagnosticată cu aniridie (indiferent dacă este izolată sau face parte din sindromul WAGR), se recomandă următoarele investigații:
Managementul în cazul unui copil diagnosticat cu sindromul WAGR cuprinde:
Sindromul Wilms sau WAGR se caracterizează prin prezența unor manifestări sistemice asociate aniridiei: nefroblastom – aniridie – anomalii genito-urinare – retard mintal, manifestări determinate de deleția cromozomului 11p13. Cunoscut anterior sub numele de Miller, sindromul WAGR apare sporadic și include aproximativ o treime din pacienții diagnosticați cu aniridie.
Aniridia izolată poate fi cauzată de diferite anomalii cromozomiale: deleții care afectează întreaga regiune PAX 6 sau o parte a acesteia, translocații și inversii care perturbă unitatea de transcripție a genei. În legătură cu nefroblastomul, Knudson a propus un model genetic care condiționează apariția tumorii în funcție de două mutații: în formele unilaterale, sporadice se bănuiește că sunt determinate mutații postzigotice iar în formele ereditare, prima mutație ar fi moștenită (prezentă în celule germinale la părinți), transmisibilă, prezigotică, iar a doua a doua mutație se produce postzigotic. Această teorie nu a fost demonstrată la urmașii pacienților cu afectare bilaterală. La copiii care asociază aniridie și nefroblastom a fost identificată o deleție a brațului scurt al cromozomului 11 (11p13) care include gena supresoare tumorală WT1.
Aceasta este cea mai frecventă tumoră renală malignă a copilului; diagnosticul cât mai precoce este foarte important deoarece prezintă o mare sensibilitate la radioterapie și chimioterapie.
Sindromul WAGR este determinat de deleția cromozomului 11p13 care duce la haploinsuficiență genelor PAX 6 și WT1. În 50% din cazuri a mai fost descoperită a treia genă implicată, gena BDNF. Această genă este localizată pe cromozomul 11p14.1 și codifică factorul de creștere GAP – 43 care joacă un rol în creșterea și dezvoltarea neuronilor și realizarea conexiunilor sinaptice încă din stadiul embrionar. Astfel, gena BDNF are rol în funcționarea normală a sistemului nervos; deleția acesteia fiind cauza apariției redardului mintal în sindromul WAGR. Dizabilitatea intelectuală (definită ca IQ < 74) se observă la 70% dintre pacienți, iar tulburările de comportament includ: hiperactivitatea cu deficit de atenție (ADHD), autism, anxietatea, depresia și tulburarea obsesiv – compulsivă.
Copiii care prezintă manfestări clinice caracteriste sindromului WAGR trebuie testați citogenetic pentru indentificare a anomaliei cromozomiale (deletie 11p) care include genelor PAX6 și WT1. Cel mai frecvent este folosit testul FISH. Odată pus diagnosticul de sindrom WAGR, pacinetul este supus investigațiilor de specialitate în vederea evaluării prognosticului și a metodelor de tratament.
| Testele genetice de diagnostic al sindromului WAGR | ||
| Afecțiunea | Gena implicată | Testare genetică |
| Aniridie | PAX 6 | Analiză ADN (secvențiere) |
| Sindrom WAGR | PAX 6, WT1 | FISH |
Odată pus diagnosticul de sindrom WAGR, pacinetul este supus investigațiilor de specialitate în vederea conduitei terapeutice:
Sindromul Gillespie este caracterizat prin triada: aniridia parțială, ataxia cerebelară non – progresivă și dizabilitatea intelectuală. Se deosebește de aniridia clasică prin faptul ca este o afecțiune cu transmitere autozomal recesivă (cealaltă transmițându-se pe cale autozomal dominantă) și reprezintă doar 2% din cazurile de aniridie. Primul caz de sindrom care asociază aniridie, retard mintal și ataxie cerebeloasă a fost raportat de către Gillespie în 1965 în cazul a doi copii, frate și soră a căror părinți erau sănătoși și neconsangvini. Sora în vârstă de 22 de ani prezenta irisul redus la un rest de țesut, marginile festonate, mișcările oculare normale și absența nistagmusului. Alte manifestări clinice asociate au fost dizabilitatea intelectuală, proba indice-nas modificată iar reflexele osteo- tendinoase erau de asemeni modificate.
Gena implicată în apariția sindromului Gillespie a fost recent identificată și se numește gena ITPR1 care codifică proteina IP3R1. În sistemul nervos central, gena IP3R1 este exprimată abundent și formează homo- sau heterotetramerii împreună cu genele IP3R2 și IP3R3, omoloage care acționează ca și canale de eliberare a ionilor de calciu în reticulul endoplasmatic.
Manifestările neurologice asociate cu mutațiile genei ITPR1 au fost inițial observate la șoareci și ulterior descoperite la om. S-a demonstrat că absența genei ITPR1 provoacă ataxie, distonie, multiple mișcări anormale și moarte timpurie. Imagistica prin rezonanță magnetică (RMN) evidențiază frecvent atrofie cerebeloasă, adesea proeminentă, afectând în principal vermusul. Relativ recent, pacienții cu sindrom cerebelos, dizabilități intelectuale și aniridia au fost considerați parte componentă a sindromul Gillespie.
Marien și colaboratorii săi au efectuat un studiu pe 2 familii în care 7 membri prezentau sindromul Gillespie, iar din punct de vedere al manifestărilor neurologice s-au constatat diferite manifestări neurologice prezentate în tabelul de mai jos.
| Manifestări neurologice prezente în sindromul Gillespie | ||
| Manifestarea neurologică | Proporția prezentă la pacienți | |
| Nistagmus | Prezent la toți subiecții investigați | |
| Ataxie cerebeloasă | Prezentă la toți subiecții investigați | |
| Tremor postural | Prezent la 5 din 7 persoane | |
| Dizartrie | Prezent la 3 din 7 persoane | |
| Hipotonia generală | Prezent la toate persoanele investigate | |
| Semne piramidale | Absente | |
| Semne extrapiramidale | Prezente la 6 din 7 persoane | |
| Clonus | Prezent la 4 din 7 persoane | |
| Dismetrie | Prezentă la 6 din 7 persoane | |
| Neuropatie periferica | Absenta | |
| Crize de epilepsie | Absente | |
| Retard intelectual | La 2 persoane-sever, la 4 persoane-mediu, | |
Diagnosticul sindromului Gillespie are la bază testarea genetică și cuprinde analiza cromozomială (cariotip) și testare FISH pentru deleții sau duplicații, sau secvențierea genei ITPR1; de asemenea analiza arborelui genealogic este indicată a se realiza.
În ceea ce privește diagnosticul diferențial, în literatura de specialitate este menționat faptul că un defect al irisului asemănător cu cel din sindromul Gillespie se întâlnește și în cadrul sindromului de disfuncție a musculaturii netede sistemice care apare în urma substituției argininei din poziția 179 în cadrul genei ACTA2. Analizele funcționale ale acestor două proteine în țesutul iris nu sunt raportate.
Aniridia va fi explorată și manageriată în departamentul de oftalmologie. Tratamentul este simptomatic și vizează în special fotofobia, nistagmusul, astigmatismul sau strabismul. Ataxia va fi explorată în departamentul de neurologie, unde pacientul beneficiază de examenul clinic și investigațiile paraclice imagistice ca de exemplu rezonanța magnetică nucleară la nivel cerebral.
Pe lângă testul IQ (care în general se situează între 70, 80, ajungând uneori și la 90 sau 95) se mai fac anumite teste care dovedesc gradul de afectare neurologică: de exemplu scrierea de mână după dictare arată un scris tremurat, cu litere mari, de amplitudine inegală. În figura 2.9 se poate observa testul scrierii de mâna după dictare.

Un alt test cu valoare orientativă îl reprezintă testul vizuo – constructiv sau examenul desenului spontan, copiat sau indicat se realizează prin utilizarea de figuri geometrice sau desene simple. Pune în evidență erori legată de controlul inadecvat al direcției de mișcare, al forței, al vitezei și al amplitudinii. De asemenea, se mai testează: vorbirea repetată, vorbirea automată, studiul exprimării orale, studiul înțelegerii limbajului scris și studiul tulburărilor de calcul.
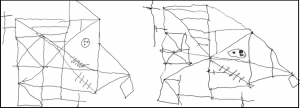
Pe lângă investigații și tratament, copiii diagnosticați au nevoie de terapie și consiliere psiho – terapeutică în vederea integrării în societate.
În ceea ce privește sfatul genetic, sindromul Gillespie este moștenit în mod autosomal recesiv, părinții unui individ cu această afecțiune poartă fiecare câte o copie a genei mutante, dar de obicei nu prezintă semne și simptome. Riscul unei noi sarcini cu un copil afectat este de 25%.
Ai nevoie de consultații oftalmologice? Îți recomandăm optica medicală Cristalin Optik.

Cataracta , Conjunctivita , Dezlipirea de retina Keratoconus

Cataracta , Conjunctivita , Dezlipirea de retina Glaucom

Accident ischemic tranzitor , Accident vascular cerebral (AVC) , Cefaleea - durerea de cap Epilepsia
Hipertensiunea arteriala (HTA) , Gastroenterita , Pielonefrita acută Lupusul eritematos sistemic - LES
Site-ul NewsMed.ro se adresează oricărei persoane care prezintă interes cu privire la subiecte din sfera medicală şi care decide să nu rămână nepăsătoare atunci când vine vorba de asigurarea propriei sănătăţi.
© Copyright 2025 NewsMed - Toate drepturile rezervate.








