Căutare
Căutare
Toate procedurile
Analiza detaliată
Analiza detaliată
Osteomielofibroza, cunoscută şi sub denumirea de mielofibroză idiopatică sau mielofibroză primară sau de mielofibroză cu metaplazie mieloidă este o boală mieloproliferativă cronică având la bază proliferarea clonală a celulei stem pluripotente şi caracterizată prin coexistenţa a două procese:
Aceste procese au loc atât în măduva osoasă cât şi în teritoriile extramedulare (splină, ficat, alte teritorii). Existenţa acestor tulburări explică evoluţia bolii în două etape: una de mieloproliferare (panmieloză şi metaplazie mieloidă extramedulară) şi alta de insuficienţă medulară.
Mielofibroza idiopatică poate fi o afecţiune primară (ca şi componentă a grupului neoplaziilor mieloproliferative cronice Philadelphia negativ), dar ea poate reprezenta şi etapa finală a evoluţiei altei boli mieloproliferative (leucemie mieloidă cronică, policitemia vera, trombocitemia esențială).
Osteomielofibroza este o boală rară (aproximativ 5 cazuri noi la 1.000.000 locuitori pe an), boala este mai frecventă la vârste mai înaintate (vârstă medie de 65 ani). Sunt afectate celulele stem pluripotente care proliferează excesiv.
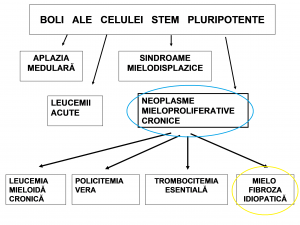
Încadrarea osteomielofibrozei în grupul afecțiunilor limfoproliferative cronice
Caracteristici comune ale neoplaziilor mieloproliferative cronice
|
În stadiul de mieloproliferare, după diferenţierea celulei stem pluripotente în celule hematopoietice şi stromale, hematopoieza medulară va determina o panmieloză, iar cea extramedulară metaplazia mieloidă în splină, ficat, limfoganglioni, alte teritorii.
Celulele stem CD34 pozitive patologice, ca şi unii precursori mai maturi, sunt mobilizaţi din teritoriile medulare şi colonizează de la început teritorii extramedulare, în principal splina şi ficatul (hematopoieză extramedulară). Metaplazia mieloidă nu este secundară fibrozării măduvei osoase, ci debutează concomitent cu mieloproliferarea. Apare splenomegalia voluminoasă în care, pe lângă proliferarea celulară, are loc şi o liză a elementelor sanguine. În sângele periferic hematopoieza extramedulară este indicată de prezenţa eritrocitelor în picătură (dacriocite) sau cu forme bizare, fragmentate, a fragmentelor de megacariocite, de apariţia de precursori celulari eritro-leuco-trombocitari (eritroblaşti, formulă leucocitară deviată la stânga).
Insuficienţa medulară se produce în urma dezvoltării procesului de fibroză medulară. Acesta apare de la începutul bolii, este preponderent în măduva osoasă şi determină scăderea progresivă a ţesutului hematopoietic. Geneza procesului fibroproliferativ nu este pe deplin elucidată. O serie de produşi (citokine, chemokine) care stimulează proliferarea fibroblaştilor (cu secreţie crescută de colagen) sau care inhibă activitatea colagenazelor şi împiedică distrugerea colagenului, sunt eliberaţi din megacariocitele patologice, displazice sau în urma unor interacţiuni celulare care implică monocitele, trombocitele, granulocitele. Printre aceste citokine se numără factori de creştere megacariocitari (transforming growth factor beta, platelet-derived growth factor), endoteliali (epidermal growth factor), fibroblastici (basic fibroblast growth factor), interleukina 1 şi alţii.
Neoangiogeneza medulară (formarea de vase sanguine, în principal capilare, adesea anarhice, în teritoriile tumorale) este un fenomen frecvent observat în toate bolile mieloproliferative cronice, ducând la o marcată creşetere a densităţii microvasculare medulare. În mielofibroza primară, în particular, procesul de neoangiogeneză este observat atât la nivelul măduvei cât şi în teritoriile cu hematopoieză extramedulară. Apariţia şi dezvoltarea acestui fenomen este promovat de factori de creştere endoteliali, citokine, chemokine, metaloproteinaze din matricea extracelulară.
Mieloproliferarea clonală este acompaniată, în mielofibroza primară, de un sindrom inflamator secundar, generat de un nivel ridicat al unor citokine proinflamatorii. Secundar acestuia, apar modificări ale stromei medulare (fibroza, osteoscleroza, neoangiogeneza), mobilizarea în circulaţie a progenitorilor CD34 pozitive; unele citokine (interleukine, IL – 8, IL – 10, IL – 15, IL – 2) par să deţină rol în evoluţia clonală a bolii. Nivelul plasmatic crescut al citokinelor proinflamatorii este incriminat în geneza simptomelor constituţionale de boală (scădere ponderală, febră, prurit, transpiraţii).
Dereglările genetice care determină apariţia celulei stem clonale (patologice) nu sunt cunoscute. Dezvoltarea subsecventă a mutaţiei JAK2V617F (şi a altora) va oferi clonei neoplazice un marcat avantaj de proliferare, permiţându-i să scape de sub controlul mecanismelor fiziologice reglatoare. Nu se cunoaşte încă clar de ce prezenţa aceleiaşi leziuni genetice (JAK2V617F) determină fenotipuri diferite de neoplazie mieloproliferativă cronică – PV, TE sau MP. Sunt implicaţi probabil mai mulţi factori, printre care starea de homo sau heterozigot pentru JAK2V617F, încărcătura alelică a mutaţiei, expresia ei pe diferiţi precursori (eritroizi, megacariocitari, granulocitari), implicarea altor leziuni genetice, epigenetice, moleculare. Studiile (genetice, de biologie moleculară) privind neoplaziile mieloproliferative cronice au cunoscut, în ultimul deceniu, o mare amploare. Se speră clarificarea mecanismelor care duc la apariţia şi dezvoltarea acestor boli pentru ca, în final, să fie posibilă utilizarea unor mijloace terapeutice eficiente şi personalizate.
Debutul mielofibrozei primare este insidios, necaracteristic şi constă în oboseală, inapetenţă, pierdere ponderală, discomfort abdominal cu senzaţie de apăsare în hipocondrul stâng. Mai pot fi prezente dureri osoase (îndeosebi în membrele inferioare) sau un sindrom hemoragipar moderat (epistaxis, gingivoragii, purpură).
Examenul clinic – obiectiv al pacientului cu osteomielofibroză este dominat de splenomegalia marcată. În momentul diagnosticării bolii, splina este de obicei de volum considerabil, ajungând în cursul evoluţiei la mărimi gigante. Ea este dură, netedă şi nedureroasă. Hepatomegalia este aproape întotdeauna prezentă. Hipertrofia hepato – splenică poate provoca diverse sindroame de compresie de vecinătate. Paloarea asociată cu discret icter (traducând un proces de hemoliză) se accentuează progresiv. În 50% din cazuri anemia este prezentă încă de la diagnosticarea bolii. Ea este severă în faza de insuficienţă medulară (mieloscleroză), când la producerea ei concurează mai mulţi factori: dislocarea eritropoiezei, hematopoieza splenică insuficientă şi ineficientă, hemoragiile şi deficitul de folaţi.
Pe lângă anemie se constată o creştere a numărului leucocitar prin granulocitoză absolută. Niciodată însă numărul leucocitar nu depăşeşte 50.000/mmc (discordanţa dintre o splenomegalie enormă şi un număr leucocitar doar moderat crescut trebuie să orienteze diagnosticul înspre o mielofibroză primară). În evoluţie numărul leucocitelor poste deveni scăzut. Numărul trombocitar este variabil pe parcursul evoluţiei bolii.
Aspectul frotiului de sânge periferic este caracteristic şi poate sugera diagnosticul, fiind prezent tabloul hematopoiezei extramedulare: morfologia eritrocitară este modificată, poikilocitoza fiind foarte exprimată prin apariţia hematiilor în „lacrimă”, în formă de „pară” sau ovală, până la aspecte bizare. Sunt prezenţi eritroblaşti şi forme tinere din seria granulocitară (tablou leuco-eritroblastic), precum şi macrotrombocite şi fragmente de megacariocite. Pe lângă devierea spre stânga (până la mieloblast) se constată neutrofilie, eozinofilie şi bazofilie.
După o primă fază hipercelulară, măduva devine hipoplazică. Puncţia sternală se execută, de obicei, cu dificultate din cauza îngroşării zonei corticale a osului. Destul de frecvent, puncţia aspiratoare este „albă”, sau sucul medular foarte redus, hipocelular.
Diagnosticul poate fi confirmat prin biopsie osteo – medulară şi examenul histopatologic din fragmentele prelevate. Secţiunile histologice pun în evidenţă creşterea ţesutului fibros, cu benzi scleroase, între care, pot fi găsite insule de măduvă hematopoietică activă. In aceste regiuni megacariocitele sunt mai numeroase, neregulate, hipolobulate; de asememnea se observă distensia sinusoidelor medulare, neoangiogeneză şi hematopoieză intravasculară.
Mutaţii importante pentru diagnostic, dar care nu au relevat influenţă prognostică semnificativă:
Mutaţii a căror prezenţă implică influenţă prognostică:
Diagnosticul pozitiv de mielofibroză primară se precizează în prezenţa celor trei criterii majore și două dintre criteriile minore:
Criterii Majore |
Criterii minore |
|
|
Tot în cadrul diagnosticului, o etapă importantă este și realizarea unui diagnostic diferențial. Acesta se face cu:
Evoluţia bolii este, de obicei, lentă. Starea bolnavului poate fi bună o perioadă lungă de timp, cu producerea treptată a insuficienţei medulare. Au fost descrise forme acute, cu o evoluţie doar de câteva luni, şi forme cronice (de la 3 la 15 ani). La un număr de cazuri (20% în primii 10 ani de evoluţie) mielofibroza primară se transformă în leucemie acută mieloblastică. Pe lângă aceasta, insuficienţa medulară (mai ales infecţiile intercurente) reprezintă principala cauză de deces.
Supravieţuirea pacienţilor este dependentă de numeroşi factori. Cea mai utilizată este stratificarea DIPPS-plus (Dynamic International Prognostic System). Factorii care implică un impact prognostic negativ sunt: vârsta > 65 ani, valoarea hemoglobinei < 10 g / dl, leucocite > 25.000 / mm3, trombocite < 100.000 / mm3, blaşti circulanti >/= 1%, dependenţa de transfuzii, prezenţa simptomelor constituţionale (scădere ponderală, febră, transpiraţii), cariotip nefavorabil.
Pacienţii sunt consideraţi a avea risc prognostic:
Mielofibroza primară este este în continuare o boală incurabilă. Tratamentul metaplaziei mieloide cu mielofibroză este în general paliativ şi trebuie adaptat fazei evolutive a bolii şi grupei de risc prognostic.
Singurul tratament cu potenţial curativ este transplantul de celule stem hematopoietice. Este aplicabil doar în puţine cazuri, având în vedere riscurile asociate procedurii (mortalitate şi morbiditate ridicată) şi vârsta în general înaintată a pacienţilor. Intră în discuţie ca mijloc terapeutic în cazul pacienţilor tineri (< 55ani), în grup de risc ridicat sau intermediar-2 sau în prezenţa leziunilor moleculare „de risc” (pozitivitate ASXL1 şi/sau negativitate CALR), care au donator compatibil. Asigură o supravieţuire mediană la 5 ani de aproximativ 50%, cu o rată a recurenţei bolii în jur de 30% la 5 ani.
Frecvenţa lor va fi dată de amploarea simptomelor date de anemie şi nu de valorile hemoglobinei; pentru a evita încărcarea organismului cu fier, la pacienţii politransfuzaţi se administrează şi tratament chelator de fier (Desferaxirox).
Hidroxicarbamida (Hidroxiuree) – doze mici (500 – 100 mg pe zi) – atenuează proliferarea megacariocitară şi acţiunea factorilor de favorizare a fibrozei medulare, poate reduce dimensiunile splinei, leucocitoza, trombocitoza, ameliorează simptomele datorate sindromului hipercatabolic. Eficientă mai ales în cazurile JAK2 V617F pozitive.
Talidomida, Lenalidomida (indicată în cazurile cu del5q), Pomalidomida (la cazurile JAK2V617F pozitive, fără splenomegalie marcată). Influenţează angiogeneza şi acţiunea citokinelor proinflamatoare (IL – 6, TNF), stimulează proliferarea şi activitatea celulelor natural – killer, a limfocitelor T, producţia de gamma interferon şi IL – 2.
Ruxolitinib (Jakavi) – este un inhibitor neselectiv JAK1/JAK2, ce acţionează atât pe formele mutante JAK2 V617F cât şi pe cele nemutante (eficient deci şi în formele JAK2 V617F negative). Nu determină abolirea sau reducerea semnificativă a clonei maligne medulare JAK2 V717F, are efect minor asupra fibrozei medulare. Are însă acţiune inhibitorie asupra celulor neoplazice din splină (cu reducerea splenomegaliei) şi prin diminuarea citokinelor proinflamatorii, ameliorează simptomele generale (scăderea ponderală, febra, pruritul, transpiraţiile). Este aprobat pentru tratamentul mielofibrozei primare şi a mielofibrozei post – policitemia vera sau trombocitemia esențială din 2011 în SUA şi din 2012 în Europa; în România se utilizează din 2014.
Mulţi alţi inhibitori ai căii de semnalizare intracelulară JAK/STAT sau downstream faţă de aceasta sunt aflaţi în faza de studiu: inhibitori JAK2 (Momelotinib, Lestaurtinib, Pacritinib, etc), inhibitori Hsp90 – heat-shok protein 90 (Panobinostat), inhibitori PI3K/mTOR (Everolimus), inhibitori ai kinazei PIM, inhibitori MEK, etc.
Aceasta este indicată în cazurile cu splenomegalie gigantă, cu fenomene compresive, infarcte repetate, hipersplenism confirmat sau hipertensiune portală cu varice esofagiene; este necesară însă existenţa unei hematopoieze restante corespunzătoare. Morbiditatea perioperatorie şi postoperatorie este ridicată (peste 25% din cazuri) şi include infecţii, tromboze ale venelor abdominale, sângerări; uneori apare hepatomegalia tumorală progresivă prin infiltrare de boală, trombocitoza marcată. Sunt păreri conform cărora splenectomia ar putea grăbi apariţia fazei blastice (transformarea în leucemie acută).
Focarele de hematopoieză extramedulară (peritoneu, pleură, limfonoduli, coloană vertebrală, piele, etc) pot fi iradiate cu doze mici (100 – 1000 cGy în 5 – 10 şedinţe). Splenomegalia gigantă, cu contraindicaţii pentru splenectomie, poate beneficia de radioterapie paliativă în doze mici (100 cGy în 5 – 10 şedinţe), cu riscul citopeniilor severe. Durerile osoase pot fi ameliorate prin iradierea 100 – 400 cGy teritoriilor dureroase. Hipertensiunea pulmonară secundară mielofibrozei primare se poate ameliora prin iradierea pulmonară cu doze mici, 100 cGy.
Aceasta se face cu Allopurinol și hidratare intravenoasă.
Se recomandă în faze avansate de boală, în prezenţa ascitei refractare, a sângerărilor variceale recurente. Șuntul porto – sistemic intrahepatic transjugular reprezintă o variantă de şunt porto-sistemic neselectiv, calibrat, prin care se creează un şunt intraparenchimatos între o venă suprahepatică şi ramul drept al venei porte. Accesul implică un radiolog intervenţionist, care instalează o proteză metalică expandabilă între cele două vase, folosind un abord transjugular. Tehnica nu este nici simplă şi nici rapidă, necesitând o aparatura performantă şi o echipă antrenată. Principala problemă a şunturilor intrahepatice este durata de funcţionare, afectată de stenozare şi trombozare. Jumătate din TIPS-uri sunt nefunţionale la un an. În aceste condiţii, se înţelege că scăderea presiunii portale prin acest procedeu nu este definitivă, fiind folosit cu predilecţie în aşteptarea transplantului hepatic. Fiind un şunt neselectiv complicaţia majoră este reprezentată de encefalopatia portală, care poate afecta majoritatea pacienţilor.

Accident ischemic tranzitor , Accident vascular cerebral (AVC) , Cefaleea - durerea de cap Epilepsia
Hipertensiunea arteriala (HTA) , Gastroenterita , Pielonefrita acută Lupusul eritematos sistemic - LES
Site-ul NewsMed.ro se adresează oricărei persoane care prezintă interes cu privire la subiecte din sfera medicală şi care decide să nu rămână nepăsătoare atunci când vine vorba de asigurarea propriei sănătăţi.
© Copyright 2025 NewsMed - Toate drepturile rezervate.


")




