Căutare
Căutare
Toate procedurile
Analiza detaliată
Analiza detaliată
Micozisul fungoid (fiind cunoscut și sub numele de sindrom Alibert – Bazin) face parte din categoria limfoamelor maligne și se caracterizează printr-o proliferare cutanată a limfocitelor T. Acesta este întâlnit mai ales la persoanele în vârstă, fiind mai frecvent la bărbaţi decât la femei (în raport de 2 la 1).
Schema clinică de sistematizare a limfoamelor maligne |
|
Linia celulară B |
Linia celulară T şi NK |
I) Limfoame indolente (cu risc scăzut):
Extralimfonodular (limfomul MALT cu limfocite B)
|
I) Limfoame indolente (cu risc scăzut):
|
II) Limfoame agresive (cu risc intermediar):
|
II) Limfoame agresive (cu risc intermediar):
|
III) Limfoame foarte agresive (cu risc ridicat):
|
III) Limfoame foarte agresive (cu risc ridicat):
|
|
|
Limfocitele T reprezintă limfocite cu un rol central în imunitatea mediată celular. Ele diferă de alte limfocite, cum ar fi celulele B sau celulele NK (natural killer), prin prezența unui receptor pentru celulele T, pe suprafața membranei celulei. Ele sunt numite celule T, deoarece acestea se maturizează în timus. Există mai multe subseturi de celule T, fiecare cu o funcție specifică.
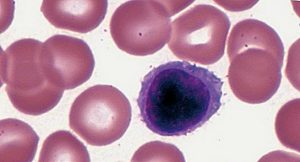
Toate limfocitele T provin din celulele stem hematopoietice din măduva osoasă. Ulterior, aceste celule derivate din celule stem hematopoietice migrează în timus, unde se divid rapid, generând o populație mare de timocite imature. Pe măsură ce acestea se maturizează, timocitele devin dublu – pozitive (CD4+ și CD8+), și în cele din urmă, devin limfocite mature, dar naive imunologic mono – pozitive: CD4+ și CD8- = limfocite T helper) sau CD4- și CD8+ = limfocite T citotoxice, care din timus ajung în țesuturile periferice. Aproximativ 98% din timocite mor în timpul procesului de maturizare din timus, nefiind in stare sa treacă de selecțiile pozitive sau negative, în timp ce cele 2% ramase devin celulele T imunocompetente.
Timusul contribuie cu mai puține celule odată cu înaintarea în vârstă. Odată cu micșorarea treptată, cu aproximativ 3% pe an, a timusului la adult, există o scădere corespunzătoare a producției de limfocite T naive, crescând rolul expansiunii celulelor T periferice în protecția imunitară a persoanelor în varstă.
Deşi cauza bolii nu este cunoscută, sunt incriminaţi ca factori de risc precum: expunerea la substanţe chimice, stimularea antigenică cronică, infecţii fungice şi virale ale pielii, hipersensibilitatea la expunerea la soare, aglomerarea neoplazică familială (îndeosebi limfoame şi leucemii). În unele cazuri, a fost confirmată infecția cu virusul HTLV – I (Human T–Lymphotropic Virus). Celulele care proliferează sunt limfocite T cu tropism dermo – epidemic foarte pronunţat aparţinând subsetului de limfocite T helper şi care circulă în mod preferenţial la nivelul pielii (în prezent, epidermul este considerat a face parte din sistemul imun: celulele Langerhans din epiderm fac „prezentarea” antigenului, iar keratinocitele secretă un număr de citokine imunoreglatoare).
Bolnavii cu micosis fungoid se prezintă cu antecedente de dermatită cronică sau „eczemă”, cu evoluţie trenantă, progresivă şi cel mai adesea însoţită de prurit supărător. Din punct de vedere clinic, boala evoluează în trei etape și anume:
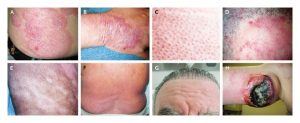
Iniţial, micosisul fungoid se manifestă doar prin manifestări cutanate. Acestea sunt sub formă de pete, placarde şi tumori. Petele cutanate eritematoase pot avea 2 – 6 cm diametrul şi apar cel mai adesea pe o anumită zonă a corpului, îndeosebi pe trunchi (abdomenul inferior, fese, coapse). Uneori au caracter atrofic, pielea este foarte subţire, prezintă aspecte pigmentare sau teleangiectazii, apar mai frecvent pe membre, torace şi pot avea peste zece cm în diametru. Aceste leziuni sunt însoţite de un prurit moderat şi adenopatiile sunt absente.
Infiltratele cutanate sub formă de placarde de 2 – 4 cm diametrul, alteori mai extinse, sunt de culoare roşiatică sau brună, pot apare pe orice zonă a corpului şi sunt însoţite, într-un grad mai accentuat, de prurit. Uneori pot fi sub forma unor papule. În cazurile cu placarde infiltrative extinse pot fi depistate adenopatii.
Progresia bolii va duce la un stadiu tumoral al acesteia, în care apar noduli fermi, roşiatici, de 1-3 cm, îndeosebi la nivelul scalpului şi al feţei. În acest stadiu mai pot apare zone infiltrate cu caracter ulcerat.
În timp, se conturează o tendinţă la confluare a leziunilor cutanate descrise, cu diseminare în ganglionii regionali, iar ulterior în diverse organe (plămâni, splină, ficat, rinichi). Din punct de vedere clinic, apare o eritrodermie generalizată (aspectul de „om roşu”) şi ulceraţii în plăci.
Într-o formă înaintată se produce eritrodermie generalizată cu invadarea leucemică. Pruritul este extrem de intens, eritrodermia îmbracă aspect descuamativ, palmele şi plantele sunt eritematoase, uscate, hiperkeratozice şi cu fisuri dureroase. Sindromul Sezary poate apare în urma evoluţiei lente a micosisului fungoid, sau (mai rar) se poate dezvolta de novo.
Examinarea esenţială pentru clarificarea diagnosticului este biopsia cutanată care va evidenţia trei elemente și anume:
Leziunile sunt mai evidente la nivelul placardelor sau al nodulilor. Un anumit grad de fibroză este prezent în dermul papilar contribuind (alături de infiltratul limfocitar) la producerea hiperkeratozei.
Diagnosticul nu poate fi pus decât prin biopsia cutanată. El este sugerat de leziunile cutanate eritematoase, pruriginoase şi de descuamarea foarte intensă. În privinţa adenopatiilor existente, trebuie făcută diferenţierea între aspectul reactiv (limfadenopatia dermatopatică) şi invadarea limfomatoasă ganglionară (doar acest ultim aspect trădând o fază avansată a bolii).
Stadializarea bolii se face similar cu cea a tumorilor solide (sistemul TNM). Pentru afirmarea diagnosticului de sindrom Sezary este necesară prezenţa în sângele periferic a unui procent de cel puţin 15, de celule cu aspect morfologic identic celui descris în leziunile cutanate.
Evoluţia micosisului fungoid este lentă, în funcţie de extinderea leziunilor cutanate şi de afectarea ganglionară, dar cu variaţii de la caz la caz. În formele cu afectarea unui teritoriu cutanat restrâns se poate ajunge la o supravieţuire de 5 – 10 ani, în cele cu eritrodermie generalizată de 1 – 3 ani, pe când în cazul sindromului Sezary aceasta este în jur de un an de zile.
Tratamentul micosisului fungoid este în funcţie de stadiul bolii şi cuprinde un tratament local (topic) şi un tratament general:
Site-ul NewsMed.ro se adresează oricărei persoane care prezintă interes cu privire la subiecte din sfera medicală şi care decide să nu rămână nepăsătoare atunci când vine vorba de asigurarea propriei sănătăţi.
© Copyright 2025 NewsMed - Toate drepturile rezervate.







