Căutare
Căutare
Toate procedurile
Analiza detaliată
Analiza detaliată
Sindroamele poliglandulare autoimune, cunoscute și sub denumirea de “patologie endocrină autoimună multiplă” sunt afecţiuni relativ rare, care asociază afecţiuni endocrinologice, neurologice, dermatologice, gastroenterologice ş.a., având în comun o afecțiune autoimună. Patologia endocrină se poate manifesta ca hipofuncție sau hiperfuncţie a glandei endocrine interesate. Coexistenţa cu diverse boli autoimune non – endocrine este posibilă.
Primele descrieri ale unor cazuri care prezentau sindroame poliglandulare autoimune datează de peste un secol şi jumătate. În anul 1855, Thomas Addison a descris primul caz cu insuficienţă corticosuprarenaliană idiopatică, anemie pernicioasă şi vitiligo. Studiul sindroamelor poliglandulare autoimune a luat o amploare mare în ultimii ani, ca urmare a aprofundării cunoştinţelor în domeniul patologiei autoimune. Acest lucru a permis identificarea pacienţilor în stadii subclinice şi iniţierea precoce a terapiei de substituţie hormonală. Descoperirea unor noi autoanticorpi şi corelarea lor cu manifestările clinice a contribuit la înţelegerea apariției bolilor şi la punerea la punct a unor metode noi de tratament. Identificarea de noi autoantigene a permis conceperea unor teste noi, ce facilitează un diagnostic cât mai precoce. Progresele înregistrate în desluşirea acestor sindroame vor contribui, probabil, la o mai bună cunoaştere a sistemului imun şi a anomaliilor acestuia, ceea ce ar putea constitui baze ale descoperirii unor terapii preventive eficiente.
Glanda endocrină (hipofiza, tiroida, paratiroidele, glandele suprarenalele, pancreasul endocrin și gonadele – testiculul şi ovarul) este un organ care fabrică unul sau mai multe produse de secreţie, pe care le eliberează direct în sânge. Sistemul endocrin reglează reproducerea, creşterea şi dezvoltarea, homeostazia organismului, producţia, depozitarea şi utilizarea energiei prin intermediul unor substanţe (hormoni) eliberate din organe specializate (glande endocrine). Hormonii, odată sintetizaţi, sunt eliberaţi în circulaţie acţionând asupra celulelor “ţintă”, fie la distanţă de locul secreţiei (acţiune endocrină), fie în vecinătatea locului unde au fost sintetizaţi (acţiune paracrină).
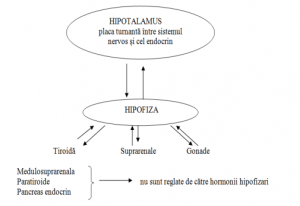
Ierarhizarea sistemului endocrin
Autoimunitatea se referă la faptul că organismul nostru nu îşi mai recunoaşte ca proprii structuri din componenţa sa (de exemplu, celule, ţesuturi, părţi din celulă etc.) şi le “atacă” prin formarea de anticorpi, ceea ce duce la perturbarea echilibrului organismului, cu apariția bolilor autoimune (un exemplu clasic al autoimunității este lupusul eritematos sistemic).
Sindromul poliglandular autoimun tip 1 |
→ Pentru diagnostic sunt necesare cel puţin două afecţiuni. |
Sindromul poliglandular autoimun tip 2 |
|
Sindromul poliglandular autoimun tip 3 |
|
Sindromul poliglandular autoimun tip 4 |
|
În clinică, cea mai des folosită încadrare a sindroamelor poliglandulare autoimune recunoaşte două subtipuri majore și anume: sindromul poliglandular autoimun tip 1 și sindromul poliglandular autoimun tip 2. Practic, s-a renunțat la sindroamele sindromul poliglandular autoimun tip 3 și sindromul poliglandular autoimun tip 4, actualmente fiind descrise două tipuri clasice – sindromul poliglandular autoimun tip 1 sau forma juvenilă, mai rară și sindromul poliglandular autoimun tip 2 sau forma adultă, mult mai frecventă. La aceste două mari sindroame se mai adaugă câteva forme particulare pe care le-am menționat mai sus, care sunt extrem de rar întâlnite.
Aceste două subtipuri de sindroame poliglandulare autoimune diferă atât în ceea ce priveşte afecţiunile componente, cât şi particularităţile modului de apariție. Anumite manifestări clinice sunt comune ambelor sindroame (boala tiroidiană autoimună, boala celiacă). Din punct de vedere clinic, sindroamele poliglandulare autoimune sunt precedate de o fază latentă asimptomatică, caracterizată de prezenţa autoanticorpi specifici unei afecţiuni, care reprezintă markerii bolii. Anumite afecţiuni componente sunt asimptomatice (boala celiacă) și altele simptomatice, dar tardiv diagnosticate (boala Addison, anemia pernicioasă, ş.a.).
Sindromul poliendocrinopatiei autoimune tip 1, sindrom cunoscut şi ca APECED (Autoimmune Polyendocrinopathy – Candidiasis – Ectodermal Distrophy) sau poliendocrinopatia autoimună juvenilă sau sindromul Whitaker este o afecțiune foarte rară și are o transmitere autosomal recesivă, caracterizată de o distrucţie autoimună la nivelul mai multor ţesuturi, deficit parţial al autoimunităţii mediate celular şi distrofie ectodermală. Sindromul este indus de mutaţii ale genei autoimune reglatoare AIRE de pe brațul lung al cromozomului 21.
Sindromul poliendocrinopatiei autoimune tip 1 este destul de rar. Se estimează că incidenţa ar fi mai mică de un caz la 100.000 de locuitori pe an. Acest sindrom este extrem de rar în Statele Unite ale Americii, fiind mai frecvent în Europa, în anumite comunități cu un grad mare de consanguinitate (la evrei iranieni – un caz la 9.000 de locuitori; în Finlanda – un caz la 14.000 de locuitori; în Sardinia – un caz la 25.000 de locuitori; în Norvegia – un caz la 80.000 de locuitori). Raportul sex feminin versus sex masculin este de 0,8 – 2,4 la 1, existând o oarecare preponderență a sexului feminin. Sindromul poliglandular autoimun tip 1 se asociază cu morbiditate şi mortalitate semnificativă, în general similară fiecărei componente a acestui sindrom. Ambele pot fi reduse prin depistarea precoce a sindromului și prevenirea sau tratarea situațiilor care pun în pericol viața acestor pacienți, cum ar fi criza addisoniană sau criza de tetanie.
Sindromul poliglandular autoimun tip 1 este o boală cu transmitere autosomal recesivă. Gena sindromului, numită AIRE („autoimmune regulator gene”) este localizată pe braţul lung al cromozomului 21 şi a fost identificată în anul 1997. Mutaţia R257X a acestei gene este responsabilă de majoritatea cazurilor de sindrom poliglandular autoimun tip 1. Prezenţa concomitentă a alelei HLA DQB1 conferă protecţie împotriva diabetului zaharat tip 1. Până în prezent au fost descrise peste 70 de mutaţii, majoritatea fiind transmise autozomal recesiv. Gena AIRE este exprimată mai ales în timus, unde joacă un rol important în inducerea toleranţei celulelor T. Mutaţiile genei AIRE induc sinteza unor proteine modificate, răspunzătoare de distrucţia autoimună a organelor ţintă, secundar afectării toleranţei imunologice a pacienţilor.
Candidoza cronică este determinată de imunodeficienţa celulelor T. Hipoparatiroidismul se instalează secundar acţiunii autoanticorpilor specifici citotoxici (anti – celule paratiroidiene şi / sau anti – receptor pentru calciu). Boala Addison este indusă printr-un mecanism similar, prin intermediul auticorpilor anti 21-hidroxilază). Se bănuiește că procesul distructiv autoimun este mediat de celulele T. Bolnavii dezvoltă o gamă largă de autoanticorpi faţă de proteinele prezente în organele afectate (adesea enzime intracelulare). Autoanticorpii reprezintă markeri diagnostici și care preced instalarea manifestărilor clinice.
Diagnosticul de certitudine al sindromul poliglandular autoimun tip 1 presupune depistarea mutaţiilor genei AIRE şi dozarea anticorpilor anti – interferon. Bolnavii care prezintă cel puţin două dintre afecţiunile majore (candidoză cronică, hipoparatiroidism, boala Addison) deţin aproape întotdeauna mutaţii AIRE.
Entităţile majore ale sindromului poliglandular autoimun tip 1, respectiv candidoza cronică, hipoparatiroidismul şi boala Addison debutează cel mai frecvent în primii 20 ani de viaţă. În schimb, bolile asociate minore pot deveni manifeste clinic mult mai tardiv, uneori chiar în decada a-V-a de viață. Apariţia determinărilor majore recunoaşte cronologia: candidoza cronică → hipoparatiroidism → boala Addison. De obicei, candidoza cronică apare înainte de 5 ani, hipoparatiroidismul înainte de 10 ani, iar boala Addison anterior vârstei de 15 ani. După cum s-a arătat, diagnosticul sindromului poliglandular autoimun tip 1 se poate stabili atunci când pacientul prezintă cel puţin două dintre cele trei entitati majore. Triada completă apare în 50% din cazuri până la 20 de ani și în 55% până la 30 de ani.
Candidoza cronică poate prezenta diverse forme clinice cu severitate variabilă și localizări multiple. Uneori pot apărea carcinoame epiteliale cu celule scuamoase la nivelul mucoasei bucale, a limbii şi esofagului, sau infecţii severe prin asplenismul asociat (septicemii fulminante). Se mai descriu: candidoză intestinală (asimptomatică sau simptomatică) şi vulvo – vaginită. În general, infecţia nu afectează mai mult de 5% din suprafaţa dermică. Candidoza generalizată a fost comunicată la bolnavii aflaţi sub medicaţie imunosupresivă.
Sindromul poliglandular autoimun tip 1 poate interesa și alte glande endocrine, cum ar fi tiroida sau gonadele. Cele mai frecvente afectări sunt, însă, cele menționate deja – hipoparatiroidismul şi boala Addison. Hipogonadismul hipergonadotrop afectează predominant populaţia feminină, care prezintă: amenoree primară, afectarea sexualizării pubertare sau menopauză precoce. Boala tiroidiană autoimună este dominată de hipotiroidie. Diabetul zaharat tip 1 nu depăşeşte 18%, iar insuficienţa hipofizară poate interesa unul sau mai multi tropi (cel mai adesea fiind hormonul de creștere).
Sindromul poliglandular autoimun tip 1 asociază concomitent și mai multe componente non – autoimune, specifice. Distrofia ectodermală încadrează: hipoplazia smalţului dentar, kerato – conjunctivita şi defecte ale unghiilor. Pot să apară și alte afectări autoimune non – endocrine, cum ar fi: gastrita cronică atrofică şi anemia pernicioasă, disfuncţia intestinală cu malabsorbţie, hepatita autoimună, nefropatia tubulo – interstiţială, vitiligo, alopecie ş.a.
Asplenia (lipsa splinei) se asociază cu prognostic foarte sever uneori. În determinismul acesteia sunt implicate cauze autoimune şi vasculare. Asplenia compromite răspunsul autoimun faţă de bacteriile încapsulate (de exemplu, pneumococ). Suspiciunea de asplenie este ridicată de prezenţa corpilor Howell – Jolly, trombocitoză, anizocitoză, poikilocitoză etc.
Diagnosticul pozitiv se stabileşte, de obicei, prin evidenţierea a cel puţin două dintre cele trei afecţiuni de bază: candidoza cronică, hipoparatiroidismul sau boala Addison. Diagnosticul de sindrom poliglandular autoimun tip 1 va fi suspectat la indivizi cu vârsta sub 30 de ani, care dezvoltă candidoză muco – cutanată asociată cu o boală autoimună endocrină. Evaluarea ulterioară va include: o anamneză amănunţită, examen clinic complet şi teste de screening pentru imunopatiile cunoscute ca asociate sindromului poliglandular autoimun tip 1.
Insuficienţa paratiroidană este susţinută de modificările metabolismlui fosfo – calcic, ce relevă calcemie scăzută, fosforemie crecută, calciurie şi fosfaturie scăzute. Concentraţia parathormonului este scăzută, iar investigaţiile imunologice pot evidenţia prezenţa anticorpilor anti – paratiroidieni în 11 – 38% dintre cazuri.
Insuficienţa corticosuprarenaliană determină următoarele modificări umorale: hiponatremie şi hiperpotasemie, hipoglicemie, anemie cu eozinofilie şi limfocitoză. Explorările hormonale evidenţiză cortizolemie bazală, aldosteron plasmatic şi dehidroepiandrosteron scăzute şi ACTH (hormonul adrenocorticotrop) crescut. Investigaţiile imunologice pot evidenţia prezenţa anticorpilor anticorticosuprarenalieni (anti 21-hidroxilază) la cel puţin 81% dintre pacienţi, în unele serii fiind raportată prezenţa lor la 100% din cazuri în momentul diagnosticului.
Autoanticorpii faţă de diverse antigene organ – specifice pot fi detectaţi cu mult timp înaintea apariţiei semnelor clinice. Identificarea autoanticorpilor organ – specifici oferă şi informaţii referitoare la cauza și mecanismul de apariție al bolii. Sensibilitatea şi specificitatea autoanticorpilor pentru apariția bolii clinice este apreciată la 60 – 80%. Determinările imunologice vor interesa şi consanguinii (consanguini de gradul unu, doi şi trei). În condiţiile unei suspiciuni justificate de sindromul poliglandular autoimun tip 1 este necesară testare genetică pentru evidenţierea mutaţiilor genei AIRE.
Tratamentul sindromului poliglandular autoimun tip 1 se adresează afecţiunilor individuale pe care le prezintă pacientul. Terapia bolii constă în: substituţie hormonală, tratamentul candidozei şi al bolilor asociate (asplenism).
Monitorizarea bolnavilor sau a consanguinilor impune o atitudine de expectativă faţă de alte boli autoimune. De aceea, după stabilirea diagnosticului de sindrom poliglandular autoimun tip 1, bolnavii vor fi atent monitorizaţi atât în ceea ce privește tratamentul, cât şi pentru decelarea unor boli autoimune adiţionale.
Insuficienţa corticosuprarenaliană acută sau criza addisoniană este consecinţa secreţiei inadecvate de hormoni glucocorticoizi şi mineralocorticoizi, care se manifestă cel mai frecvent prin colaps cardio – vascular, pierdere de sare şi hipoglicemie. Incidența bolii Addison în populație este de 4,7 – 6,2 cazuri la 1.000.000 locuitori. Până în anii 1940 etiologia insuficienţei corticosuprarenaliene primare a fost predominant tuberculoza, ulterior pe primul loc (circa 90% din cazuri) aflându-se etiologia autoimună. 25% din insuficienţele corticosuprarenaliene primare nu sunt diagnosticate până la criza addisoniană.
Insuficienţa corticosuprarenaliană acută poate fi primară (datorată distrugerii morfologice şi consecutiv funcţionale a cortexului suprarenalian – în boala Addison), secundară (hipopituitarism global sau deficit izolat de ACTH – adenocorticotrop) sau terţiară (afectare hipotalamică). Afectarea primară interesează în general tot cortexul, antrenând un deficit secretor nu doar al glucocorticoizilor ci şi al mineralocorticoizilor şi al sexoizilor suprarenalieni. Din această cauză manifestările clinice sunt mai severe şi, uneori, întrucât instalarea insuficienţei corticosuprarenale primare este insidioasă, debutul aparent să se facă printr-o criză addisoniană. Insuficienţa corticosuprarenală acută secundară afectează preponderent glucocorticoizii. Persistenţa secreţiei de mineralocorticoizi face criza acută mai puţin probabilă. La un pacient cunoscut, criza adrenală apare în urma unor afecţiuni severe pe fondul unei substituţii inadecvate – fie subdozaj, fie lipsa de adaptare a dozei în condiţii de stres.
a) La nou – născut și sugar:
b) La adult și copil:
În faza de prodrom a bolii, se observă: astenie, adinamie, melanodermie (în boala Addison), hipotensiune arterială.
Criza adrenală este dominată de hipotensiune arterială, dar tabloul clinic este mult mai complex datorită, pe de o parte, asocierii altor semne şi simptome ale insuficienţei corticosuprarenale, cum ar fi cele digestive, pe de altă parte simptomelor datorate etiologiei acutizării insuficienţei corticosuprarenaliene: sepsis, traumatism, hemoragie hipofizară sau corticosuprarenală.
Durerile abdominale şi febra pot duce la confuzia cu abdomen acut chirurgical, o intervenţie chirurgicală fără protecţie cu glucocorticoizi putând fi fatală !!
Manifestări clinice sugestive pentru criza addisoniană sunt:
|
Explorările de laborator se prelevă odată cu puncţia venoasă necesară instituirii tratamentului (nivelul cortizolului plasmatic și cortizolul liber urinar, cortizolul salivar sodiu, potasiu, rezerva alcalină, glicemie, uree, hemoleucogramă). Tratamentul se începe imediat, fără a aştepta confirmarea diagnosticului !! Modificări electrolitice sunt: hiponatremie (88 % cazuri), hiperpotasemie (64 % cazuri), hipercalcemie (< 30 %), hipoglicemie (66 %) – de intensitate variabilă, uneori foarte gravă, azotemie. Modificări hematologice: anemie, eozinofilie.
Investigațiile hormonale în criza addisoniană sunt:
Odată stabilit diagnosticul de certidudine este bine de făcut un diagnostic diferențial cu: abdomenul acut chirurgical, sindroame hemoragice de diverse cauze, meningoencefalite, septicopioemii, alte come – diabetică, hipoglicemică, uremică.
Obiectivele tratamentului sunt:
Monitorizarea tratamentului trebuie realizată ca în orice urgenţă și include: tensiunea arterială, frecvenţa cardiacă, temperatura, diureza, volumul pierderilor digestive, teste biologice (ionogramă, uree, creatinină, glicemie, hemoleucogramă), hemoculturi; urocultură.
Se face cu soluţie izotonă (glucoză 5%, normosalină – NaCl 0,9%):
Hemisuccinat de Hidrocortizon:
Dexametazonă – dacă este accesibilă, nu modifică dozarea cortizolului plasmatic. Se adminstrează 4 mg intravenos în bolus.
Cortizon acetat – administrare intramuscular: nu este util în formele acute (absorbţie încetinită din cauza colapsului), dar este util după revenirea tensiunii arteriale, favorizând scăderea dozei adminstrate intravenos.
Dacă nu există un factor major precipitant, după 1 – 3 zile se poate trece la administrarea de glucocorticoizi per os (respectându-se scăderea treptată a dozei până se ajunge, în 5 – 6 zile, la doza substitutivă cronică de 15 – 25 mg Hidrocortizon sau 5 – 10 mg Prednison / zi).
Nu sunt necesar câtă vreme doza de hemisuccinat de Hidrocortizon este mai mare de 100 mg/zi. Cortizolul are afinitate pentru receptorii mineralocorticoizi dar în mod normal e inactivat renal, prin transformare în cortizon sub acţiunea 11 beta-hidroxi-dehidrogenazei. Când cantitatea de glucocorticoizi este mare (de exemplu, sindrom Cushing endogen sau exogen sau, în cazul nostru, în substituţia din criza adrenală) cortizolul nu e total inactivat şi activează receptorul mineralocorticoid determinând efecte mineralocorticoide majore. După scăderea dozei de glucocorticoizi, la doza substituivă de glucocoritoiczi se adaugă: Fludrocortizon (Astonin, Fluorinef) 0,05 – 0,2 mg/zi per os.
O serie de investigaţii (urocultură, hemoculturi, teste de coagulare) sunt necesare pentru identificarea causei precipitante. Febra, fără decelarea unei infecţii, poate fi datorată hipocortizolemiei. Antibioterapia este indicată doar în febrele care persistă după echilibrare cortizolică. Hemoragia se tratează prin repleţie volemică cu plasmă sau sânge.
În condiţiile instituirii prompte a terapiei pacientul îşi revine în primele 24 ore; absenţa ameliorării stării clinice în primele 3 ore pune în discuţie cauza decompensării corticosuprarenale.
Tratamentul substitutiv normal se administrează oral: 15 – 20 mg hemisuccinat de Hidrocortizon sau 5 – 7,5 mg Prednison, divizat în 2 – 3 prize. Aceste doze sunt uşor suprafiziologice, dar supradozarea este probabil necesară din cauza inactivării rapide a cortizolului în cortizon în urma ingestiei. Doza de mineralocroticoizi este 0,05 – 0,2 mg Fludrocortizon pe zi.
Atât pacientul cât şi familia trebuie să beneficieze de informaţii detaliate privind afecţiunea, modul de manifestare, riscurile, terapia şi măsurile preventive. O brăţară sau un card pe care să se menţioneze tratamentul cronic cu cortizon trebuie să se afle mereu asupra sa, astfel încât în urgenţă să poată fi. Este util ca pacientul să aibă în permanenţă asupra sa o trusă de urgenţă care să conţină un glucocorticoid injectabil. În caz de decompensare sau de accident, administrarea a 100 mg hemisuccinat de hidrocortizon îi asigură protecţia în timpul transportului la spital.
Pacientul trebuie să ştie să îşi adapteze doza în funcţie de circumstanţă:
Sindromul poliendocrinopatiei autoimune tip 2 reprezintă cea mai frecventă formă de patologie endocrină autoimună. Se estimează că prevalenţa este de un caz la 20.000 de locuitori, iar incidenţa ei de aproximativ unul – două cazuri la 10.000 de locuitori pe an, adică de 10 – 20 de ori mai mare decât a sindromului poliendocrinopatiei autoimune tip 1. În realitate, aceasta este mai mare dacă se iau în calcul şi cazurile cu boală subclinică. Sindromul poliglandular autoimun tip 2 este mai frecvent la femei (raportul sex feminin versus sex masculin este de 3 – 4 la 1) și prezintă incidenţa maximă în decadele II – VI de viață, putând fi comun mai multor generaţii.
Modul de apariție al sindromului nu este pe deplin elucidat. Sindromul poliglandular autoimun tip 2 este o afecţiune genetică mai complexă, controlată de haplotipul HLA, implicat în diverse boli autoimune. Complexul genic HLA este localizat pe cromozomul șase şi cuprinde trei clase (I, II și III). Alelele clasei II, HLA – DQ, HLA – DR şi într-o mai mică măsură HLA – DP sunt cei mai importanţi determinanţi ai afecţiunilor din sindromul poliglandular autoimun tip 2, care codează anumite proteine ale complexului major de histocompatibilitate, localizate la nivelul celulelor care prezintă antigenul.
Sindromul poliglandular autoimun tip 2 tinde să dețină agregare familială. Modul de transmitere al sindromului este poligenic, autosomal dominant, cu penetranţă incompletă. Un rol important îl au polimorfismele genelor sistemului HLA, localizate pe braţul scurt al cromozomului 6.
Factorii de mediu pot avea rol declanşator al procesului imun. Cu toate că au fost identificaţi unii dintre aceştia, în majoritatea cazurilor de sindrom poliglandular autoimun tip 2 nu se poate evidenţia prezenţa unui trigger (factor declanșator). Principalii factori de mediu cu rol în apariția unor componente ale poliendocrinopatiei sunt: gliadina pentru boala celiacă, metimazol pentru autoanticorpii insulinici, penicilamina pentru miastenia gravis, rubeola congenitală şi ingestia precoce de lapte de vacă, cazeină sau grâu pentru diabetul zaharat tip 1, anticorpii monoclonali anti – CD52 folosiţi în tratamentul sclerozei multiple, pentru boala Basedow Graves, interferonul pentru hipotiroidism. Dezvoltarea unei determinări autoimune la bolnavii cu sindrom poliglandular autoimun tip 2 presupune atât o predispoziţie genetică, cât şi pierderea toleranţei.
Diferenţe privind mecanismul de apariție între sindromul poliglandular autoimun tip 1 și 2 |
|
| Sindromul poliglandular autoimun tip 1 | Sindromul poliglandular autoimun tip 2 |
| Debut în copilărie | Debut în viaţa adultă |
| Mutaţii ale genei AIRE | Fără mutaţii ale genei AIRE |
| Fără asocieri cu haplotipul HLA | Asociere cu HLA – DR3, DR4 |
| Imunodeficienţă prezentă | Fără imunodeficienţă |
| Candidoză muco – cutanată prezentă | Fără candidoză muco – cutanată |
Autoanticorpii reprezintă markeri serologici ai procesului imun aflat în derulare la nivelul glandelor endocrine şi prezic apariţia ulterioară a manifestărilor clinice. Unii dintre aceştia, însă, au şi rol patogenetic. Este vorba despre anticorpii anti – acetilcolină pentru miastenia gravis şi anti – receptor TSH pentru boala Basedow Graves. În general, autoanticorpii se leagă de suprafaţa celulei cu sau fără a avea influențe funcţionale. Prevalenţa autoanticorpilor este în general mare la debutul clinic al endocrinopatiei autoimune. În timp, titrele acestora scad sau chiar se negativează. Detectarea anticorpilor organ – specifici oferă indicii privind cauza şi selectează cazurile care vor dezvolta sindromul poliglandular autoimun.
Cele mai importante autoantigene la bolnavii cu sindromul poliglandular autoimun |
|
| Autoantigene | Boală autoimună |
| Tiroperoxidază , Tireoglobulină | Tiroidita Hashimoto |
| Receptorul pentru TSH, Tiroperoxidază | Boala Basedow Graves |
| Receptorul pentru calciu | Hipoparatiroidism |
| 21 – hidroxilaza | Boala Addison |
| Decarboxilaza acidului glutamic (GAD-65), Insulina, IA-2 (ICA-512), IA- 2β (ICA 512β, fogrina) | Diabetul zaharat tip 1 |
| ATP-aza H / K dependentă a celulelor gastrice parietale | Gastrita autoimună |
| Factorul intrinsec (celulele gastrice principale) | Anemia pernicioasă |
| Transglutaminaza, gliadina | Boala celiacă |
| Enzimele citocromului: P450 – D6, C9, A2 | Hepatita autoimună |
| Tirozinaza | Vitiligo |
Atât aspectele clinice, cât şi datele de laborator sunt similare celor din bolile izolate care fac parte din tabloul sindromului poliglandular autoimun tip 2. Manifestările clinice autoimune (endocrine, sistemice) din boală prezintă o mare variabilitate, în funcţie de fiecare caz în parte.
Prevalenţa diverselor afecțiuni imunologice în sindromul poliglandular autoimun tip 1 și 2 |
||
Afecțiune autoimună |
Sindromul poliglandular autoimun tip 1 (%) |
Sindromul poliglandular autoimun tip2 (%) |
| Candidoză muco – cutanată | 76 – 100 | 0 |
| Boală Addison | 72 – 100 | 0 – 100 |
| Hipoparatiroidism | 86 – 93 | rar |
| Boală tiroidiană autoimună | 2 – 18 | 69 – 88 |
| Diabet zaharat tip 1 | 2 – 23 | 23 – 52 |
| Hipogonadism hipergonadotrop | 17 – 72 | 3,6 – 10 |
| Hepatită autoimună | 13 – 20 | 3 |
| Anemie pernicioasă | 13 – 31 | 1 – 2 |
| Sindrom diareic cronic | 18 | 4 – 12 |
| Vitiligo | 8 – 26 | rar |
| Alopecie | 12 – 40 | 4,5 – 12 |
Din tabel, se poate observa că că bolnavii cu sindrom poliglandular autoimun tip 2 dezvoltă foarte rar hipoparatiroidism şi niciodată candidoză cronică. Prevalenţa bolii tiroidiene autoimune este mult mai mare în sindromul poliglandular autoimun tip 2 versus sindromul poliglandular autoimun tip 1.
Debutul diverselor entităţi se succede la intervale de câţiva ani sau decade, fiind precedat de o fază subclinică, premergătoare celei manifeste. Adesea, insuficienţa corticosuprarenaliană poate precede celelalte determinări endocrine, fiind mai frecvent asociată cu alte boli imunologice (comparativ cu diabetul zaharat tip 1 şi boala tiroidiană autoimună izolată). Boala tiroidiană autoimună se întâlneşte la 50% dintre cei cu boala Addison, în timp ce diabetul zaharat tip 1 asociat este prezent la 15% dintre pacienţii cu boala Addison. Boala tiroidiană autoimună este dominată de hipotiroidie, boala Basedow Graves fiind mult mai rară.
Sindromul poliglandular autoimun tip 2 mai poate prezenta și alte boli imunologice considerate minore: hipogonadism hipergonadotrop, vitiligo, alopecie, hepatită cronică, gastrită cronică atrofică cu sau fără anemie pernicioasă şi chiar hipofizită. Prevalenţa afecțiunilor imunologice minore în sindromul poliglandular autoimun tip 2 este mai redusă comparativ cu sindromul poliglandular autoimun tip 1.
Pentru diagnosticul sindromului poliglandular autoimun tip 2 sunt necesare: examen clinic, diverse determinări umorale, dozarea anticorpilor organ – specifici şi numeroase teste funcţionale. Practic, diagnosticul se stabileşte, de obicei, prin evidenţierea bolilor autoimune care pot să facă parte din acest sindrom. Plecând de la simptomatologia şi manifestările clinice specifice fiecărei boli se vor face explorări metabolice, hormonale şi imunologice care să confirme boala suspectată clinic.
Se ştie că persoanele cu o singură boală autoimună prezintă un risc mai mare decât populaţia generală de a dezvolta a doua boală autoimună. Mai mult, la pacienţi cu sindrom poliglandular autoimun tip 2 debutul bolilor autoimune se face în timp, de-a lungul mai multor ani. Cei mai mulţi dintre aceşti pacienţi nu prezintă insuficienţe endocrine clinic manifeste de la început. De aceea au fost concepute diverse protocoale de diagnostic precoce, chiar în stadiu preclinic, pentru celelalte suferinţe autoimune, odată ce prima boală autoimună a fost diagnosticată. Există însă controverse legate de investigaţiile ce se indică ca metode de screening la aceşti bolnavi. De exemplu, în cazul diabetului zaharat tip 1 se indică, ca un screening de rutină, explorarea funcţiei tiroidiene prin dozări hormonale.
Odată ce un pacient a fost identificat ca având sindrom poliglandular autoimun tip 2 prin decelarea a două boli autoimune endocrine sau non – endocrine se recomandă investigaţii mai complexe pentru depistarea unor alte posibile boli într-un stadiu cât mai precoce. Acestea trebuie să includă şi investigaţii imunologice complexe, în cazul în care explorările metabolice şi hormonale nu decelează prezenţa bolilor autoimune: autoanticorpi asociaţi diabetului zaharat – antigenul insular 2, anti – insulinici şi anti – GAD (decarboxilaza acidului glutamic), anticorpi anti – tiroidieni anti – tiroperoxidază şi anti – tiroglobulină, anticorpi anti 21 – hidroxilaza pentru boala Addison, anticorpi anti – transglutaminază şi anticorpi anti – enzimele citocromului P450 pentru hepatita autoimună. La aceste cazuri cunoscute ca prezentând sindrom poliglandular autoimun tip 2 se recomandă efectuarea periodică a screeningului pentru depistarea activă şi precoce a altor componente ale acestui sindrom.
Investigații diagnostice necesare în sindromul poliglandular autoimun tip 2 |
||
| Autoanticorpi față de | Teste funcționale | Screening genetic |
|
|
|
Tratamentul în sindromul poliglandular autoimun tip 2 este centrat pe identificarea şi tratamentul diverselor condiţii autoimune.
Deoarece boala Addison reprezintă cea mai severă afecţiune, terapia acesteia necesită o atenţie specială (indiferent de tipul sindromului poliglandular autoimun). Cei cu boala Addison simptomatică vor fi trataţi cu hidrocortizon sau cortizon. Dozele substitutive sunt reprezentate de aproximativ 25 mg hidrocortizon sau 37,5 mg cortizon. Substituţia mineralocorticoidă se va face cu fludrocortizon 50 – 200 μg pe zi, în doză unică. În stabilirea dozei substitutive se va ţine cont de: tensiunea arterială, potasiu seric, activitatea reninei plasmatice (cu valori în jumătatea superioară a spectrului normal). Dozele de substituţie se vor dubla sau tripla în cazul unor intercurenţe, intervenţii ş.a. Deoarece cei cu boala Addison dețin un risc important de a dezvolta alte boli imunologice, se impune screeningul pentru autoanticorpi față de insulină, antigenul insular 2 și decarboxilaza acidului glutamic (pentru diabetul zaharat tip 1) și față de transglutaminază (pentru boala celiacă).
La cei cu sindrom poliglandular autoimun tip 2 şi boală tiroidiană autoimună este obligatorie testarea funcţiei corticosuprarenalei înainte de instituirea tratamentului hipotiroidiei. Iniţierea terapiei cu tiroxină în insuficienţa corticosuprarenaliană netratată, poate precipita o criză addisoniană.
Deoarece boala tiroidiană autoimună este foarte frecventă, atât la pacienții cu diabet zaharat tip 1, cât şi la cei cu boala Addison este necesară monitorizarea periodică a TSH-ului.
Pacienţii cu sindrom poliglandular autoimun tip 1 şi sindrom poliglandular autoimun tip 2, precum şi rudele lor apropiate, trebuie monitorizaţi pe toată durata vieţii, deoarece există riscul apariţiei de noi boli endocrine. Monitorizarea constă în efectuarea periodică, la interval de câţiva ani, a examenului fizic şi a unor teste specifice, precum şi în consilierea pacienţilor şi rudelor lor în ce priveşte simptomele şi semnele precoce ale principalelor componente ale sindromului. Au fost imaginate diverse protocoale de diagnostic precoce al afecţiunilor autoimune, chiar în stadiu preclinic, o dată ce prima boală autoimună a fost diagnosticată, fără să existe un consens în ceea ce priveşte intervalul de timp la care se repetă investigaţiile.
Testele screening sunt reprezentate de:
În cazul altor elemente componente, cum este vitiligo, screeningul constă în efectuarea unui examen fizic periodic, iar pentru hipofizită şi miastenia gravis, investigaţiile paraclinice sunt ghidate de tabloul clinic prezentat de pacient. Dacă se evidenţiază corpusculii Howell Jolly, pacienţii vor fi investigaţi în vederea diagnosticării asplenismului. În cazul sindromului poliglandular autoimun tip 1, în centrele specializate, se indică analiza moleculară a genei AIRE.
Depistarea activă a pacienţilor cu sindrom poliglandular autoimun tip 1 şi sindrom poliglandular autoimun tip 2 permite un diagnostic precoce şi instituirea unei terapii adecvate, în vederea prevenirii complicaţiilor, ceea ce permite o supravieţuire mai îndelungată şi o ameliorare a calităţii vieţii acestor pacienţi.

Dislipidemia , Nefropatia diabetica , Sindromul insulinic autoimun cu hipoglicemie Neuropatia diabetica
Accident ischemic tranzitor , Accident vascular cerebral (AVC) , Cefaleea - durerea de cap Epilepsia
Hipertensiunea arteriala (HTA) , Gastroenterita , Pielonefrita acută Lupusul eritematos sistemic - LES
Site-ul NewsMed.ro se adresează oricărei persoane care prezintă interes cu privire la subiecte din sfera medicală şi care decide să nu rămână nepăsătoare atunci când vine vorba de asigurarea propriei sănătăţi.
© Copyright 2025 NewsMed - Toate drepturile rezervate.







