Căutare
Căutare
Toate procedurile
Analiza detaliată
Analiza detaliată
 Inima este un mușchi special care se contractă ritmic și continuu, pompând sângele spre întregul corp și plămâni. Este compusă din patru camere – două superioare (atriile) și două inferioare (ventriculii). Pomparea sângelui de către inimă este condiționată de un flux de semnale electrice, care traversează inima. Semnalele electrice se repetă ciclic, și fiecare ciclu produce o bătaie cardiacă. Dacă activitatea electrică a inimii este tulburată, când apar așa numitele aritmii, abilitatea inimii de a pompa adecvat poate fi afectată. Sindromul de QT lung este o boală care afectează activitatea electrică a inimii.
Inima este un mușchi special care se contractă ritmic și continuu, pompând sângele spre întregul corp și plămâni. Este compusă din patru camere – două superioare (atriile) și două inferioare (ventriculii). Pomparea sângelui de către inimă este condiționată de un flux de semnale electrice, care traversează inima. Semnalele electrice se repetă ciclic, și fiecare ciclu produce o bătaie cardiacă. Dacă activitatea electrică a inimii este tulburată, când apar așa numitele aritmii, abilitatea inimii de a pompa adecvat poate fi afectată. Sindromul de QT lung este o boală care afectează activitatea electrică a inimii.
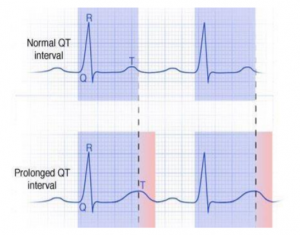
Intervalul QT măsoară o porțiune din bătaia cardiacă înregistrată pe electrocardiogramă (ECG). În timpul fiecărei bătăi cardiace un semnal electric traversează inima, inducând contracția mușchiului cardiac și pomparea sângelui. După fiecare contracție, mușchiul inimiitrebuie să aibă timp să se recupereze și să se relaxeze înainte de a primi următorul semnal electric. Durata aceasta de timp, în care are loc relaxarea, se numește interval QT. La oamenii cu sindrom de QT lung, intervalul QT este mai lung decât normal (cum sugerează și numele). Dacă următorul semnal electric ajunge prea devreme (spre exemplu, când mușchiul nu s-a recuperat complet după ultima contracție), acesta poate determina inima să bată anormal de repede, ducând la amețeală, leșin sau chiar deces. Aproximativ 1 din 2.000 de persoane au sindrom de QT lung.
Sindromul de QT lung este o boală genetică. Aceasta înseamnă că sindromul de QT lung este cauzat de un defect (o mutație) într-o genă, care poate fi transmis ereditar. Gena este o parte a ADN-ului nostru, care conține un cod pentru fabricarea unei molecule (o proteină). Orice persoană are două copii ale fiecărei gene care poate fi legată de apariția sindromului de QT lung. Acesta poate fi cauzat de mutații în genele care conțin codul pentru molecule (proteine) din inimă. O mutație în una din cele două copii ale uneia din aceste gene (de la tată sau de la mamă) este suficientă pentru a determina apariția sindromului de QT lung.
Sindromul de QT lung este o așa numită boală autozomal – dominantă și un părinte purtător are o probabilitate de 50% (1 din 2) de a transmite mutația către fiecare copil. Probabilitatea ca un copil să nu moștenească gena cu mutație este de asemenea de 50 de procente. Câteodată sindromul de QT lung poate fi o boală autosomal – recesivă. Asta înseamnă că sunt necesare mutații ale ambelor copii ale unei gene (atât de la mamă cât și de la tată) pentru a apărea sindromul de QT lung.
În funcție de gena și de mutația implicată, sindromul de QT lung va fi o boală autosomal – dominantă sau autozomal – recesivă. În unele cazuri, poate apărea o mutație nouă (de novo) în spermatozoizi, ovule sau în embrion. În aceste cazuri, părinții copilului nu au mutația și nici sindromul de QT lung, dar copilul are sindromul de QT lung și poate transmite gena mutantă copiilor săi.
Sindromul de QT lung afectează frecvent copii și adulții tineri. Cele mai frecvente simptome sunt leșinul sau colapsul. Simptomele apar adesea în timpul activităților care cresc frecvența cardiacă și nivelul de adrenalină în corp, cum se întâmplă în timpul efortului fizic (mai ales înot), situații cu încărcătură emoțională și zgomote puternice apărute brusc. Diagnosticul sindromului de QT lung poate fi dificil, deoarece mulți oameni adesea nu prezintă simptome. Totuși, când se pune diagnosticul de sindrom de QT lung, sunt disponibile terapii adecvate.
Cele mai utilizate metode diagnostice pentru sindromul de QT lung sunt: istoricul medical și familial, examinarea fizică, înregistrarea fenomenelor electrice cardiace (electrocardiograma), monitorizarea Holter și testul de efort. Din păcate, punerea diagnosticului de sindrom de QT lung poate fi foarte dificilă, deoarece mulți oameni cu aceasta afecțiune pot avea un ECG normal.
ECG-ul este o investigație de bază. Se lipesc mici plasturi (electrozi) pe piept, pe mâini și picioare. Aceștia sunt conectați prin fire cu o mașina de înregistrat ECG-ul, care timp de câteva secunde detectează activitatea electrică care formează bătăile inimii. Câteodată sunt necesare ECG-uri suplimentare sau repetate.
Testul de efort se efectuează ca și ECG-ul descris anterior, dar este înregistrat înainte, în timpul și după activitate fizică pe bandă de alergat sau bicicletă. Se detectează modificările tiparului electric al inimii apărute la efort.
Monitorizarea Holter implică utilizarea unui aparat digital de dimensiuni mici, care poate fi purtat pe o curea în jurul taliei. Patru sau șase electrozi ai aparatului se lipesc pe piept. Apoi aparatul înregistrează activitatea electrică a inimii timp de 24 – 48 de ore, sau până la 7 zile. În timpul înregistrării, toate activitățile sunt notatede către pacient într-un „jurnal” .
Aceasta este o variantă mai complicată a monitorizării Holter, descrisă mai sus. Dacă apar simptome, aparatul poate fi activat pentru a înregistra ritmul cardiac. Avantajul Cardiomemo este că nu are electrozi, astfel că poate fi aplicat pe piept în timp ce apar simptomele.
Ecocordul utilizează unde sonore, ultrasunete, pentru a vizualiza structurile cardiace. O ecografie poate detecta diverse tipuri de modificări structurale cardiace, de exemplu boli ale mușchiului cardiac sau anomalii ale valvelor inimii. Se pot identifica zonele în care mușchiul cardiac este subțiat. Pacienții cu sindrom de QT lung nu au anomalii structurale majore, dar adesea se efectuează o ecografie pentru a confirma acest fapt.
Există mai multe tipuri de sindrom de QT lung, fiecare fiind cauzat de o mutație genetică diferită. În aproximativ 70% (7 din 10) din pacienții cu sindrom de QT lung, cauza bolii poate fi depistată la nivelul genelor. La majoritatea pacienților la care se detectează o mutație, aceasta apare în una din următoarele trei gene: KCNQ1, KCNQ2 sau SCN5A. Aceste trei gene determină SQTL tip 1, 2 sau 3.
Deși nu există un remediu pentru sindromul de QT lung moștenit, tratamentele ajută în prevenirea apariției simptomelor și minimizează riscul de leșin sau stop cardiac. Tratamentul depinde de: simptome, vârstă, gen și mutația genetică specifică. Adesea este prescris un medicament numit beta – blocant, pentru a reduce aritmiile. Aceste medicamente sunt eficiente la 80 – 90% din pacienți. Beta – blocantele nu scurtează intervalul QT, dar blochează efectele adrenalinei și ale altor substanțe naturale similare la nivelul inimii, determinând scăderea frecvenței cardiace. La unii pacienți sunt necesare și alte medicamente adiționale beta – blocantelor.
Pentru pacienții la care medicamentele nu sunt eficiente sau la cei care au suferit un stop cardiac, poate fi luată în considerare implantarea unui defibrilator cardiac intern sau simpatectomia cervicală. Defibrilatorul cardiac intern poate corecta majoritatea aritmiilor amenințătoare de viață. Simpatectomia cervicală (numită și denervare cardiacă) este o procedură chirurgicală prin care se lezează nervii care eliberează adrenalina și alte substanțe naturale similare la nivelul inimii.
Medicul cardiolog va stabili frecvența cu care revine pacientul pentru controale periodice, în funcție de simptome, vârstă și tratament.
Dacă se găsește o mutație genetică la un pacient cu sindrom de QT lung, membrii familiei acestuia (începând cu rude de gradul I: mamă, tată, frați, surori și copii) pot face testul genetic într-o clinică specializată în boli cardiace genetice. Rudele la care se identifică aceeași mutație genetică sunt denumiți purtători ai mutației și vor fi urmăriți de un medic cardiolog. Rudele la care nu se identifică mutația genetică pot fi asiguraţi că nu sunt ȋn pericol. Dacă la un pacient cu sindrom de QT lung nu se găsește o mutație genetică, rudele acestuia (începând cu rudele de gradul I) sunt sfătuite să meargă la medicul cardiolog. Pacienții cu sindrom de QT lung pot avea simptome în copilărie. De aceea testarea genetică, investigațiile cardiace și tratamentul precoce pentru membrii familiei care sunt diagnosticați cu sindrom de QT lung sunt importante chiar dinprimii ani de viață.
În timpul sarcinii este important să continuați tratamentul cu beta – blocante. Câteodată este necesară schimbarea tipului de beta – blocant, deoarece nu sunt toate adecvate pentru utilizarea în timpul sarcinii. Când se iau beta – blocante în timpul sarcinii, este recomandată planificarea nașterii în spital, deoarece copilul poate avea o frecvenţă cardiacă mai scăzută. În primele nouă luni după nașterea copilului, se recomandă controale suplimentare pentru mamă deoarece în această perioadă este la risc crescut pentru apariția aritmiilor (în special, pacientele cu sindrom de QT lung tip 2).

Hipertensiunea arteriala (HTA)

Dislipidemia , Hipertensiunea arteriala (HTA) , Infarctul miocardic Insuficiența cardiacă
Anevrismele aortei toracice , Anevrismul cardiac , Anevrismul disecant al aortei Anevrismul micotic al aortei
Dislipidemia , Nefropatia diabetica , Sindromul insulinic autoimun cu hipoglicemie Neuropatia diabetica
Atac ischemic tranzitor
Boala cardiaca hipertensiva
Șocul - explicații, tipuri, manifestări clinice și tratament
Atac ischemic tranzitor , Ateroscleroza , Boala obstructivă carotidiană (Stenoza carotidiană) Diabetul zaharat si bolile cardiovasculare
Angina pectorala instabila
Hipertensiunea arteriala (HTA) , Gastroenterita , Pielonefrita acută Lupusul eritematos sistemic - LES
Hipertensiunea arteriala (HTA)
Site-ul NewsMed.ro se adresează oricărei persoane care prezintă interes cu privire la subiecte din sfera medicală şi care decide să nu rămână nepăsătoare atunci când vine vorba de asigurarea propriei sănătăţi.
© Copyright 2025 NewsMed - Toate drepturile rezervate.





")

