Căutare
Căutare
Toate procedurile
Analiza detaliată
Analiza detaliată
Sindroamele mielodisplazice sunt boli clonale ale celulei stem pluripotente caracterizate prin apariţia unei pancitopenii în sângele periferic şi printr-o măduvă osoasă hipercelulară. Din punct de vedere clinic, este vorba despre o insuficienţă medulară, iar la examenul morfologic se caracterizează prin prezenţa unor modificări displazice în una sau mai multe linii celulare în măduva osoasă sau în sângele periferic. Existenţa unui apoptoze (moarte celulară programată) crescute, în ciuda unei proliferări celulare accelerate, duce la discrepanţa dintre măduva hipercelulară şi citopenia din sângele periferic. Majoritatea pacienţilor au modificări genetice, dar incidenţa acestora depinde de metoda de diagnostic utilizată, examenul citogenetic standard fiind mai puţin sensibil decât FISH (fluorescence in situ hybridization – hibridizare fluorescentă in situ).
În populaţia generală, incidenţa sindroamelor mielodisplazice este de 5 cazuri la 100.000 de persoane, dar creşte la 50 – 60 la 100.000 peste vârsta de 70 de ani, fiind practic una dintre cele mai frecvente boli ale sângelui.
Celulele stem sunt celule nediferențiate cu un enorm potențial de diferențiere, care stau la baza dezvoltării tuturor organismelor. Acestea au un rol primordial în embriologie cât și în refacerea țesuturilor și a rănilor, sau formarea de noi celule diferențiate în organe. Celulele stem primitive generează toate tipurile de celule pe care organismele unicelulare le prezintă în cursul vieții lor (ciclul de viață).
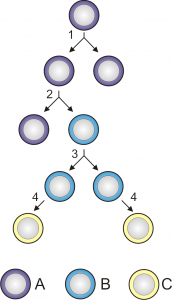
Diviziunea și diferențierea celulelor stem
(A = celulă totipotentă sau pluripotentă; B = celulă unipotentă; C = celulă diferențiată)
În funcție de potențialul lor de diferențiere, celulele stem pot fi clasificate în celule totipotente, pluripotente și unipotente. Celulele stem totipotente sunt cele mai versatile. Celulele stem pluripotente sunt asemănătoare celor totipotente în sensul că pot da naștere mai multor categorii de țesuturi dar, spre deosebire de acestea, nu pot dezvolta un întreg organism metazoar. Celulele stem unipotente sunt celulele ce dau naștere unui singur tip de celule (spre exemplu, celulele stem din stratul bazal al pielii ce dau naștere keratinocitelor).
Există trei surse accesibile cunoscute de celule stem adulte autologe la om: măduvă osoasă, țesut adipos și sânge. Celulele stem pot fi de asemenea luate din sângele ombilical imediat după naștere. Dintre toate tipurile de terapie cu celule stem, recoltarea pe cale autologică implică cel mai mic risc. Celulele stem adulte sunt frecvent utilizate în diferite terapii medicale (de exemplu, transplantul de măduvă osoasă). Celulele stem pot fi acum cultivate artificial și transformate (diferențiate) în tipuri de celule specializate cu caracteristici compatibile cu celulele diferitelor țesuturi, cum ar fi mușchii sau nervii.
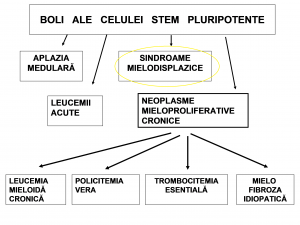
Afecțiuni ale celulei stem pluripotente
Sindroamele mielodisplazice îşi au originea în una sau mai multe mutaţii ce au loc într-un precursor tânăr al unei celule hematopoietice, sub influenţa aceloraşi factori cauzali ca în cazul leucemiilor. În afară de factorii de mediu şi / sau iatrogeni, sunt implicaţi diverşi factori de creştere celulară, în prezenţa unor pronunţate disfuncţii imunitare şi a unor importante modificări ale micromediului celular din măduva osoasă.
Iradierea şi chimioterapia pentru cancer se numără printre declanşatorii cei mai cunoscuţi ai apariției sindroamelor mielodisplazice. Pacienţii care iau medicamente chimioterapice sau cărora li se administrează tratament cu radiaţii pentru cancerele potenţial vindecabile, cum ar fi cancerul mamar sau testicular, boala Hodgkin şi limfomul malign non – Hodgkin, au riscul de a dezvolta sindroame mielodisplazice în intervalul de până la 10 ani de la tratament.
Pacienţii şi familiile acestora sunt, deseori, îngrijoraţi că sindroamele mielodisplazice ar putea fi contagioase. Nu există nicio dovadă care să sugereze că sindroamele mielodisplazice sunt provocate de un virus; de aceea, sindroamele mielodisplazice nu poate fi transmis celor dragi. Sindroamele mielodisplazice nu sunt moştenite. De fapt, se întâmplă foarte rar ca membri ai familiei, inclusiv fraţi sau surori, să fie diagnosticaţi cu sindroame mielodisplazice.
În toate sindroamele mielodisplazice, clona patologică se expansionează progresiv. În funcţie de instabilitatea acesteia, apar anomalii cromosomiale adiţionale. Leucemia acută reprezită cea mai gravă consecinţă a mutaţiilor somatice secvenţionale. Incidenţa sindroamelor mielodisplazice este în creştere, îndeosebi la persoanele în vârstă (sindroamele mielodisplazice primare). Constatarea unei mielodisplazii sub vârsta de 40 de ani trebuie să atragă atenţia mai degrabă înspre debutul unei leucemii sau înspre sindroamele mielodisplazice secundare (induse de factori mutageni: diverse substanţe chimice, citostatice alkilante sau inhibitori de topoisomerază II, radiaţii etc.).
Clona displazică are anomalii intrinseci, genetice (ale cromozomilor, ale ADN-lui, cu alterarea succesiunii normale a nucleotidelor, de obicei prin pierdere de material genetic = deleţii) sau epigenetice (hipermetilarea ADN sau acetilarea histonelor). Acestea determină anomalii ale apoptozei (moartea celulară programată).
Celula stem malignă este caracterizată printr-un potenţial de proliferare crescut, dar şi printr-o apoptoză accelerată, ceea ce duce la o maturaţie ineficientă, cu o măduvă osoasă bogată şi cu citopenie în sângele periferic. Apoptoza este mai pronunţată în stadiile timpurii ale sindroamelor mielodisplazice fiind susţinută de producerea unor citokine proinflamatorii. Sindroamele mielodisplazice apar ca urmare a unui proces desfăşurat în trepte. Micromediul medular este implicat în mecanismului de apariție al acestor sindroame prin eliberarea de factor vascular endotelial de creştere care stimulează apoptoza prin intermediul variatelor citokine şi creşte multiplicarea celulelor stem leucemice. Răspunsul imun celular mediat de limfocitele T este implicat în sindroamele mielodisplazice care apar la tineri şi care evoluează cu măduvă osoasă hipocelulară.
Tipul de sindrom mielodisplazic |
Sânge periferic |
Măduvă osoasă |
Citopenie refractară cu displazie uniliniară:
|
|
|
| Citopenie refractară cu displazie multiliniară |
|
|
| Anemie refractară cu sideroblaşti inelari |
|
|
| Anemia refractară cu exces de blaşti tip 1 |
|
|
| Anemia refractară cu exces de blaşti tip 2 |
|
|
| Sindrom mielodisplazic neclasificat |
|
|
| Sindrom mielodisplazic cu del (5q) izolată
|
|
|
Este definit de prezenţa izolată a deleţiei interstiţiale a braţului lung a cromozomului 5, fără alte modificări citogenetice asociate. Are un risc scăzut de evoluţie spre leucemie mieloidă acută, iar supravieţuirea este în jur de 9 – 10 ani atunci când procentul de blaşti din măduvă este sub 5%. Majoritatea cazurilor se încadreaza în anemia refractară. Se caracterizează prin anemie macrocitară, cu trombocitoză, leucopenia uşoară şi megacariocite mononucleare în măduvă. Numărul de precursori eritroizi este foarte scăzut (1 – 2% în măduvă). Predomină pacienţii vârstnici, de sex feminin.
Reprezintă o minoritate de sindroame mielodisplazice care evoluează cu măduvă osoasă hipocelulară (reducerea celularităţii medulare). În apariția acestor tipuri de sindroame mielodisplazice cu insuficienţă medulară este implicat un mecanism imun mediat de limfocitele T. Acestea pot să răspundă la tratamentul cu globulină anti – timocitară sau cu Ciclosporină.
Sunt reprezentate de următoarele entităţi:
Vârsta medie la diagnostic este de 70 de ani. Cea mai frecventă formă de prezentare este citopenia, de multe ori pacienţii fiind asimptomatici.
Pacienţii au în general simptome legate de anemie (paloare, dispnee) şi doar 10% au infecţii (deşi 60% au neutropenie). Infecţiile sunt mai frecvent bacteriene şi rareori virale sau fungice. Deseori febra poate fi absentă şi este prezentă doar indispoziţia, slăbiciunea. Pacienţii cu trombocitopenie pot avea echimoze şi, uneori, splenomegalie (neobişnuită în mielodisplazie, find întâlnită doar în 10 dintre cazuri). 60% dintre pacienţi au numărul de trombocite sub 150.000/mm3. Trombocitopenia izolată apare la doar 5% dintre pacienţi.
În anemia refractară și anemie refractară cu sideroblaşti inelari, evoluţia anemiei este cronică, sindromul infecţios şi hemoragic putând lipsi ani de zile. În anemia refractară cu exces de blaşti, simptomatologia este mai bogată şi numai studiul măduvei osoase poate face, în unele cazuri, deosebirea de o leucemie acută (blaşti sub 20%) sau de o anemie aplastică (măduva hipercelulară, dismielopoieza).
Leucemia mielomonocitară cronică este însoţită de splenomegalie pronunţată şi, spre deosebire de celelalte subtipuri de sindroame mielodisplazice, evoluează cu un număr leucocitar crescut (granulocitopoieza este de regulă eficientă şi numai eritropoieza şi trombocitopoieza sunt ineficiente) şi monocitoză peste 1.000/mm3.
Criteriile de diagnostic al fiecărui subtip sunt prezentate în tabelul de mai sus. Studiul frotiului periferic şi al măduvei osoase vor permite (în funcţie de procentul blaştilor şi al sideroblaştilor inelari) încadrarea în unul din subtipurile de sindroame mielodisplazice. Pentru diagnosticul leucemiei mielomonocitare cronice este necesară prezenţa a cel puţin 1.000 monocite pe mm3 în sângele periferic. De remarcat că procentul blaştilor este întotdeauna sub 20% (depăşirea acestei limite încadrează sindromul mielodisplazic într-o leucemie acută).
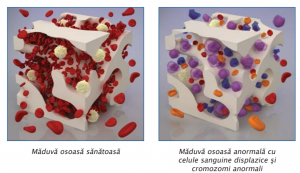
La examenul măduvei osoase, de obicei hipercelulare, sunt prezente modificări displazice pe cele trei serii celulare. O hematopoieză ineficientă (pronunţate anomalii morfologice) în prezenţa unei măduve hipercelulare, a unei pancitopenii periferice şi a elementelor sanguine mature disfuncţionale, reprezintă elementul diagnostic definitoriu.
Anomaliile morfologice cele mai importante sunt:
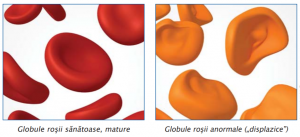
Dismielopoieza poate fi manifestată pe una, două sau pe toate cele trei serii celulare.
La analiza citogenetică, în sindroamele mielodisplazice cele mai frecvente anomalii citogenetice sunt deleţiile cromosomiale şi afectează braţul lung al cromozomilor 5, 7, 20 şi 11 şi 13 . Mai pot să apară monosomii (anomalie genetică caracterizată prin absența unmui cromozom de la nivelul unei perechi normale), trisomii (anomalie genetică caracterizată printr-un cromozom suplimentar, adică trei cromozomi în loc de perechea normală).
Diagnosticul pozitiv se pune pe baza simptomatologiei clinice şi de laborator, a aspectelor de mielodisplazie şi este confirmat prin evidenţierea unor modificări specifice. Vârsta medie la diagnostic este de 70 de ani. Cea mai frecventă formă de prezentare este citopenia, de multe ori pacienţii fiind asimptomatici.
Pentru diagnosticul sindroamelor mielodisplazice, la fel ca în leucemii acute, sunt recomandate următoarele examinări: examinarea frotiului de sânge periferic şi de aspirat medular cu coloraţie Giemsa, examinarea biopsiei osteomedulare, citochimie (peroxidaza – se poate efectua prin citometrie de flux, esteraza), imunofenotipare, examen citogenetic din măduvă (dacă măduva nu este disponibilă se poate face FISH din celule în interfază din sângele periferic) şi analiza moleculară.
Tot în cadrul diagnosticului, o etapă importantă este și realizarea unui diagnostic diferențial pentru că sindromul mielodisplazic este un diagnostic de excludere. Uneori diagnosticul nu poate fi precizat imediat şi este nevoie de dozarea unor vitamine, de probă terapeutică (de exemplu, cu vitamina B12) sau de o perioadă de observare. Trebuie diferenţiat de anemia care apare în: anemiile megaloblastice (deficitul de vitamina B12, de acid folic), anemiile congenitale diseritropoietice, anemiile sideroblastice secundare (intoxicaţii – plumb, neoplazii, boli autoimune), hipersplenismul, intoxicaţia cu alcool (produce modificări îndeosebi pe seria roşie), boli autoimune, regenerările medulare după chimioterapie, expunerea la unele antibiotice (modificările sunt tranzitorii), infecţii (HIV) boli inflamatorii, boli hepatice, cancer, anemia aplastică, mielofibroza.
Deşi sindromul mielodisplazic este privit ca o „stare preleucemică”, evoluţia este diferită în funcţie de formele acestuia. Ceva mai puţin de o treime dintre pacienţi decedează datorită infecţiilor, aproximativ o treime prin transformare în leucemie acută mieloidă, iar restul datorită comorbidităţilor legate de vârsta înaintată. Evoluţia se face din formele de anemie refractară, anemie refractară cu sideroblaști inelari sau leucemia mielomonocitară cronică spre anemia refractară cu exces de blaști sau o leucemie mieloidă acută.
Prognosticul pacienților poate fi apreciat prin utilizarea Sistemului Scorului Prognostic Internaţional (IPSS) care acordă puncte pentru anumite date:
Variabila de prognostic |
0 puncte |
0.5 puncte |
1 punct |
1.5 puncte |
2 puncte |
| % de blaşti din măduva osoasă | < 5 | între 5 și 10 | – | între 11 și 20 | între 21 și 30 |
| cariotip* | favorabil | intermediar | nefavorabil | – | – |
| citopenii | 0 sau1 | 2 sau 3 | – | – | – |
* cariotip favorabil: fără anomalii (46,XX or 46,XY), -Y, del(5q), del(20q); cariotip intermediar: prezenţa de alte anomalii, ca trisomia 8 (+8); cariotip nefavorabil: complex (3 anomalii sau anomalii ale cr 7 [7q- or -7]).
În funcţie de punctajul IPSS, se pot stabili nivele de risc (scăzut, intermediar sau crescut) care se corelează cu supravieţuirea medie a bolnavilor:
Subtipul de sindrom mielodisplazic |
Progresia spre o leucemie acută |
Supravieţuire medie |
| Anemie refractară cu sideroblaști inelari | 10% | 70 luni |
| Anemie refractară | 15% | 65 luni |
| Leucemie mielomonocitară cronică | 30% | 10 luni |
| Anemie refractară cu exces de blaști | 40% | 10 luni |
Principiile de tratament într-un sindrom mielodisplazic sunt:
|
Deseori pacienţii sunt trataţi cu acid folic şi vitamina B12, datorită modificărilor megaloblastice care pot fi prezente la examinarea aspiratului de măduvă osoasă. Alţii sunt trataţi cu preparate de glucocorticoizi pentru trombocitopenie, pentru stimularea eritropoiezei sau chiar pentru o eventuală hemoliză. La alţii se administrează vitamina B6. Toate aceste medicamente pot fi administrate pentru o perioadă scurtă, până la 2 – 3 luni, diagnosticul de sindrom mielodisplazic fiind, cum am mai menţionat, unul de excludere. Atunci când diagnosticul este însă cert, tratamentul cu aceste medicamente este considerat o greşeală (corticoterapia creşte rata infecţiilor), sau este cel puţin inutil.
Când devin simptomatici, majoritatea pacienţilor vor fi trataţi cu transfuzii regulate cu concentrat eritrocitar asociate cu tratament chelator cu fier. Transfuziile se administrează pentru menţinerea unei valori a hemoglobinei de aproximativ 9 – 10 g / dL. Necesarul de transfuzie este diferit de la un pacient la altul.
Tratamentul chelator cu fier se începe atunci când feritina creşte peste 1000 ng / mL sau când se depăşeşte un număr de 20 de unităţi de concentrat eritrocitar transfuzate. Sunt folosite trei medicamente chelatoare de fier: Desferioxamina (Desferal) este eficientă, dar necesită administrare subcutanată prelungită, fapt care reduce complianţa; Deferiprone; Deferasirox (Exjade), se administrează oral în doză de 20 mg / kg / zi şi este cel puţin la fel de eficient ca şi Desferal, dar este relativ scump.
Sub denumirea de “cel mai bun tratament suportiv” sunt grupate administrarea regulată de transfuzii, tratamentul infecţiilor cu antibiotic şi administrarea de G-CSF la pacienţii neutropenici cu infecţii.
La unii pacienţi se poate încerca stimularea eritropoiezei cu eritropoietină (eritropoietină alfa sau beta sau darbepoietina alfa). Tratamentul este eficient la cei cu eritropoietina endogenă scăzută sau în limite nromale. Asocierea de G-CSF poate determina un răspuns la pacienţi care nu au reacţionat la administrarea de eritropoteină administrată singură. Răspunsul poate fi apreciat prin creşterea valorilor hemoglobinei, reducerea necesarului de transfuzii sau chiar independenţa de transfuzii.
La acest tratament se pot asocia unul din următoarele regimuri terapeutice:
Lenalidomida, un derivat al Thalidomidei, cu efect anticitokine şi antiangiogeneză, are efect bun la pacienţii cu afectare izolată a seriei roşii, la cei cu scor IPSS scăzut şi mai ales la cei cu sindrom 5q-. Alţi inhibitori ai angiogenezei investigaţi în sindroamele mielodisplazice sunt Thalidomida, Bevacizumab şi inhibitori de receptori de tirozin kinază.
Ciclosporina în doze de 3 – 6 mg / kg corp, este eficientă în sindroamele mielodisplazice hipoplazice, mai ales la pacienţii cu genotip HLA – DQ15. Pledează pentru un mecanism predominant imun al afectării hematopoiezei la aceşti pacienţi, în care limfocitele T CD8+ au un rol inhibitor asupra hematopoiezei, la fel ca în cazul pacienţilor cu anemie aplastică (există încercări de tratament imunosupresor cu globlină anti-timocitară). 10% din pacienţii cu sindrom mielodisplazic au o boală autoimună asociată.
Singurul tratament care prelungeşte în mod cert supravieţuirea este transplantul allogen de celule stem. Doar o optime dintre pacienţi sunt eligibili pentru transplant şi doar o treime din cei transplantaţi sunt vindecaţi. Factorii de prognostic includ vârsta, durata şi starea bolii, procentul de blaşti din măduvă, anomaliile citogenetice, tipul de donor, tipul de condiţionare pretransplant. La bolnavii cu boală avansată se va administra chimioterapie pretransplant pentru a reduce numărul de blaşti.
Site-ul NewsMed.ro se adresează oricărei persoane care prezintă interes cu privire la subiecte din sfera medicală şi care decide să nu rămână nepăsătoare atunci când vine vorba de asigurarea propriei sănătăţi.
© Copyright 2024 NewsMed - Toate drepturile rezervate.





")

